
肌肉无力是重症肌无力的一个主要症状和体征,而眼外肌最易受累,是MG最常见的首发症状。目前有几种常见的MG分型系统。
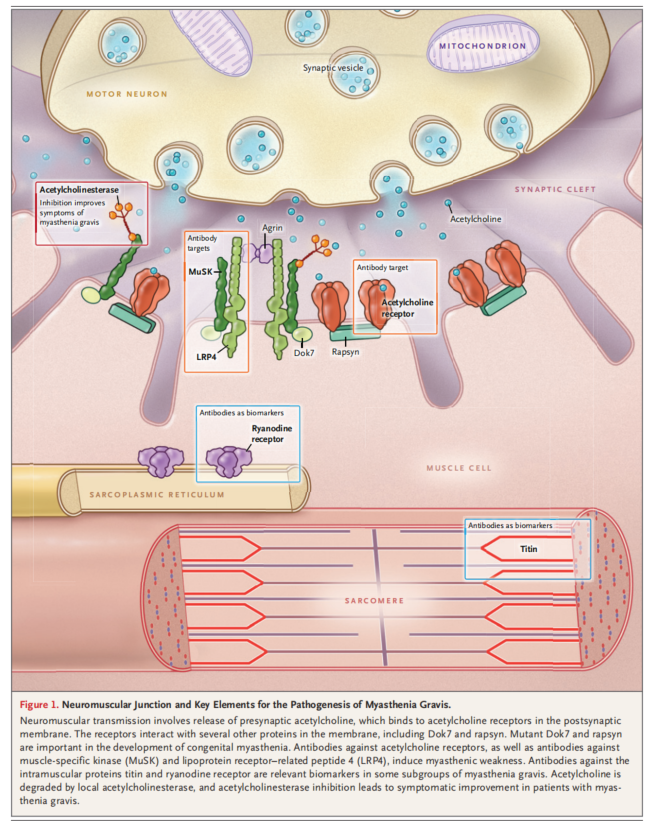
Osserman分型
1985年Osserman将成人MG根据发病年龄、受累部位、严重程度、疾病进展和预后进行分型,故称“Osserman分型”。后将Ⅱ型明确分为Ⅱa和Ⅱb两种类型,并取消肌萎缩型,形成“改良的Osserman标准”:
Ⅰ型:眼肌型(15~20%),单纯眼外肌受累。症状主要是单纯眼外肌受累,表现为一侧或双侧上睑下垂,有复视或斜视现象。肾上腺皮质激素治疗有效,预后好。
Ⅱ型:全身型,累及一组以上延髓支配的肌群,病情较I型重,累及颈、项、背部及四肢躯干肌肉群。据其严重程度可分为Ⅱa与Ⅱb型。
ⅡA型:轻度全身型(30%),进展缓慢,无危象,常伴眼外肌受累,无咀嚼、吞咽及构音障碍,下肢无力明显,登楼抬腿无力,无胸闷或呼吸困难等症状。对药物反应好,预后较好。
ⅡB型:中度全身型(25%),骨骼肌和延髓肌严重受累,明显全身无力,生活尚可自理,伴有轻度吞咽困难,时有进流汁不当而呛咳,感觉胸闷,呼吸不畅。无危象,药物敏感欠佳。
Ⅲ型:重症急进型(15%),症状危重,进展迅速,数周或数月内达到高峰,胸腺瘤高发。可发生危象,药效差,常需气管切开或辅助呼吸,死亡率高。
Ⅳ型:迟发重症型(10%),症状同Ⅲ型,从Ⅰ型发展为ⅡA、ⅡB型,经2年以上进展期,逐渐发展而来。药物治疗差,预后差。
Ⅴ型:肌萎缩型,起病半年出现肌肉萎缩,生活不能自理,吞咽困难,食物误入气管而由鼻孔呛出。口齿不清或伴有胸闷气急。因长期肌无力而出现继发性肌萎缩者不属于此型。病程反复2年以上,常由Ⅰ型或Ⅱ型发展而来。
此外还有儿童型和少年型,儿童型又分为新生儿型和先行型,大部分仅累及眼外肌,少数累及全身骨骼肌。新生儿型临床多表现为哭声低、吸吮无力,经治疗后多在一周至3个月内痊愈;先行型临床多表现为出生后短暂出现肌无力、对抗胆碱酯酶药物效果不佳,但进程缓慢。少年型多为单纯的眼外肌麻痹,部分伴有吞咽困难及四肢无力。
Osserman分型便于临床识别患者受累部位、轻重缓急和疾病进程,长期以来用作MG临床研究资料。但由于临床症状与分型关系并非绝对,存在类型转化的中间带,判断时主观性较强,故分组依据也只能作为参考。
MGFA分型
最新发布的《2020年重症肌无力诊断和治疗指南》,提出采用由美国神经病学学会(ANN)在MG基金会(Myasthenia Gravis Foundation of America,MGFA)提出的新型分型方法替代Osseman分型,旨在对疾病严重程度进行量化评估,但不能用于疗效和预后评价。

严重程度根据定量MG评分(quantitativeMG score,QMGS)评估。

compston分型
1980年Compston等根据发病年龄和胸腺病理学将MG分成3种亚型,后随着医学技术的发展,更为精确的检测标准化,最新发布的《2020年重症肌无力诊断和治疗指南》,提出重症肌无力亚组分类,指导精准化治疗,提醒临床注意其他器官并发症的出现。
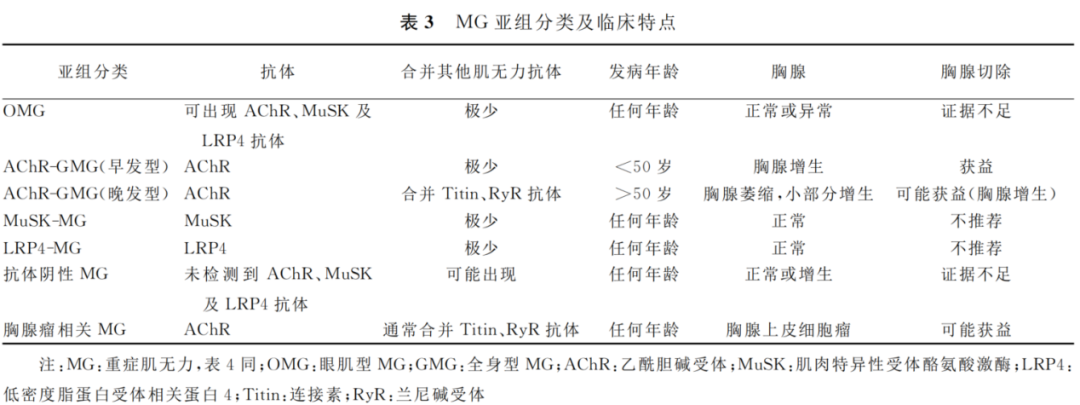
OMG:可发生于任何年龄阶段。儿童及JMG以眼肌型为主,很少向全身型转化;成人在眼肌症状出现两年内容易向全身型转化。
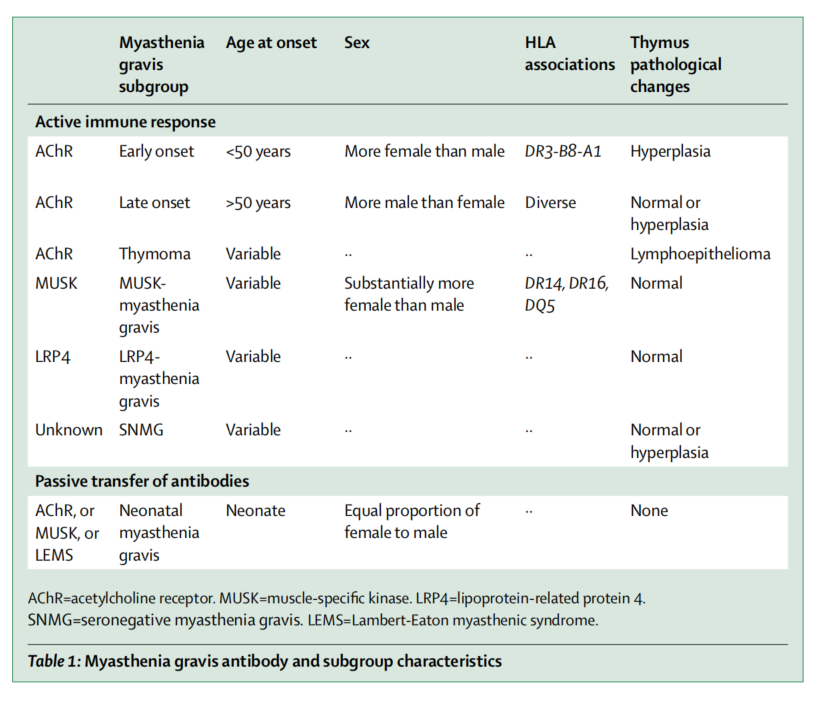
AChR-GMG:可分早晚型,以50岁为界限。早发型,女性发病略高于男性;晚发型,男性发病略高于女性。
MuSK-MG:受累肌群较局限,以球部、颈部及呼吸肌为主,其次为眼外肌、四肢肌,主要表现为球麻痹、面颈肌无力。
LRP4-MG:临床特点尚不完全明确。
抗体阴性MG:极少部分患者无上述可检测到的抗体。
胸腺相关MG:约占患者的10%~15%,属于副肿瘤综合征。
总结来说,Osserman分型操作简单、直观,但主观性强,可作为临床特征分类;MGFA分型更为简便,对患者进行问卷量化,相对较客观;Compston分型可根据不同亚群指定不同治疗方案,对患者治疗与预后有重要意义。
参考文献:
[1].NilsE. Gilhus, M.D. Myasthenia Gravis .N Engl J Med 2016;375:2570-81.
[2].Nils Erik Gilhus, Jan J Verschuuren.Myasthenia gravis: subgroupclassififi cation and therapeutic. Strategies Lancet Neurol 2015; 14: 1023–36
[3].中华医学会神经病学分会神经免疫学组.中国重症肌无力诊断和治疗指南(2020版).中国免疫学会神经免疫分会 2021,Vol.28,No.1
[4].中华医学会神经病学分会神经免疫学组.中国重症肌无力诊断和治疗指南(2015版).中国免疫学会神经免疫分会 2015,Vol.48,No.11
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#肌无力#
46
这么专业的平台,文章也很有帮助
57