
Eur J Neurol:自身抗体水平作为生物标志物在重症肌无力患者治疗中的作用
2023-04-26 医路坦克 MedSci原创
虽然重症肌无力(MG)被认为是一种免疫球蛋白G自身抗体介导的疾病,但MG自身抗体水平与疾病活动性之间的关系尚不清楚。
重症肌无力(MG)是一种神经肌肉接头(NMJ)的自身免疫性疾病,临床上以肌肉疲劳性无力为特征。它的发病率为0.3-2.8‰,每年影响全球约70万不同年龄和性别的人。这种情况通常表现为眼部症状(典型的复视和/或眼皮下垂),然后经常进展到面部、眼球、颈部、四肢和呼吸肌。
MG是由针对突触后NMJ特定分子的免疫球蛋白G(IgG)类自身抗体介导的,导致神经肌肉传递受损。在大多数患者(约85%)中,致病自身抗体针对烟碱型乙酰胆碱受体(AChRs),其他自身抗体针对肌肉特异性激酶(约6%的患者)和低密度脂蛋白受体相关蛋白4(LRP4;约2%的患者)。

AChR自身抗体(AChR-ab;主要是IgG1和IgG3亚类)致病机制包括阻断ACh结合部位,这阻碍了NMJ上ACh依赖的信号传递,随后AChR的交联、内化和随后的降解;细胞表面AChR的交联以及功能和结构的耗尽;以及突触后膜上补体的激活,导致AChR簇的破坏,膜攻击复合体的组装,以及突触后膜的破坏。麝香自身抗体(IgG4亚类的麝香抗体)抑制麝香和LRP4之间的相互作用,阻止集聚蛋白刺激的麝香磷酸化,从而破坏突触后膜结构并影响NMJ的传递。LRP4自身抗体(主要是IgG1亚类)可能的致病机制包括通过补充补体蛋白破坏突触后结构或通过减少穆斯克激活而损害信号转导。其他令人感兴趣的自身抗体的例子包括细胞内蛋白ryanodine和titin;然而,尽管它们的存在可能预示着更严重的疾病,但MG的因果关系尚不清楚。
由于自身抗体与MG的发病机制有关,其在疾病诊断和/或治疗中的临床应用具有重要的临床意义。意大利、德国和英国的指南包括自身抗体状态(例如,AChR-ab MG、Musk-ab MG、LRP4-MG、血清阴性MG)检测,作为MG患者诊断和血清学分类的公认工具。然而,除了在诊断中的这种作用外,自身抗体水平与疾病严重程度或特定临床结果之间的关系需要澄清,因为发现相互矛盾,在临床实践中的使用也是多种多样的。在一些研究中,在群体水平上,AChR自身抗体已显示出作为疾病严重程度的有效标记物,滴度与定量的重症肌无力(QMG)和美国重症肌无力基金会(MGFA)的临床分类显著相关,而最近的研究表明这种相关性不存在
综述的目的有两个:(I)调查MG患者自身抗体水平(研究报告)和疾病活动性之间的关系;(Ii)确定哪些其他因素/变量影响\“AChR-ab和Musk-ab水平/疾病活动性\”相关数据可能如何影响个体患者管理。
表1 研究选择标准
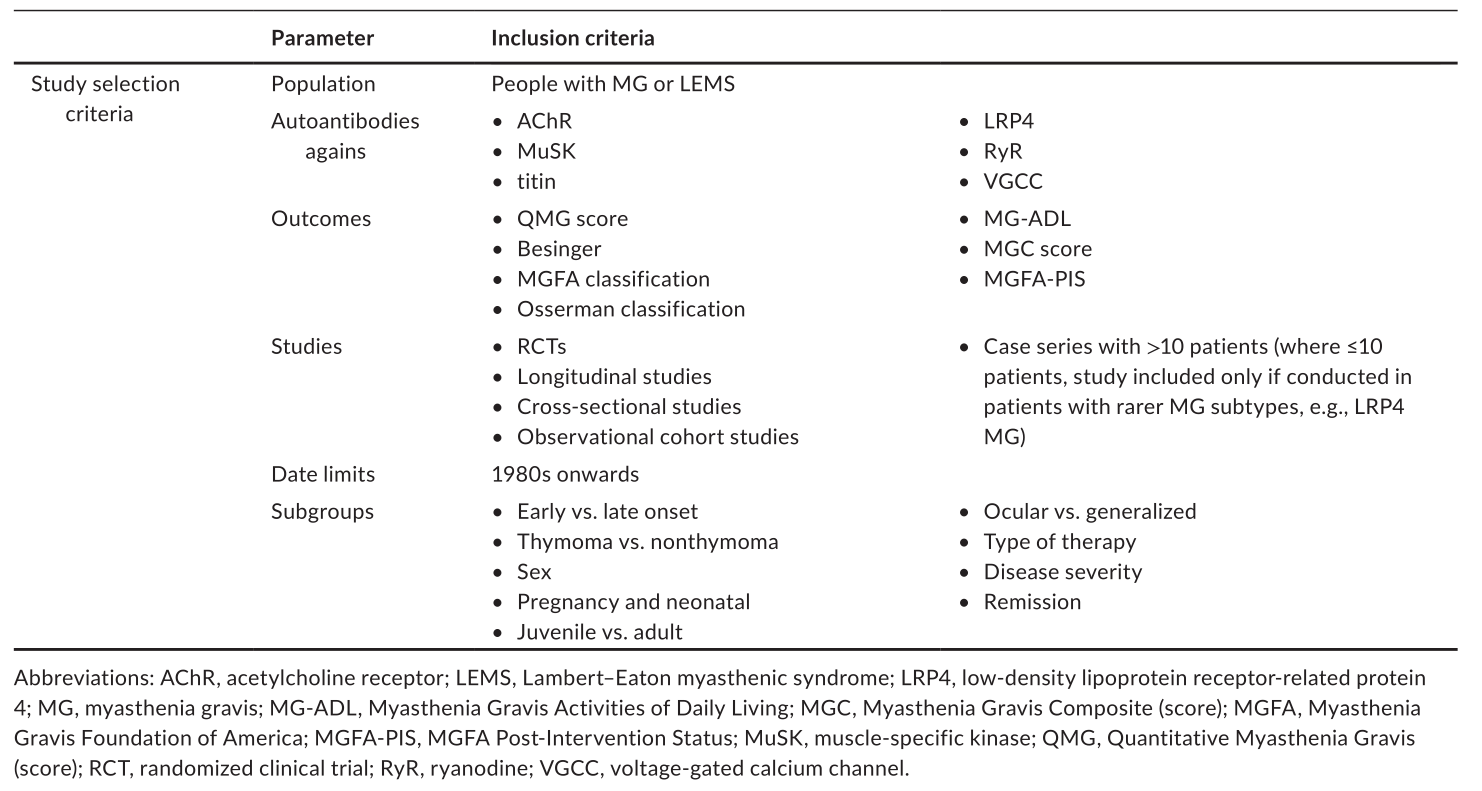
图1 PRISMA(系统综述和荟萃分析的首选报告项目):研究选择过程。乙酰胆碱受体,不包括;麝香,肌肉特异性激酶。一些研究报告了一种以上自身抗体临床结果配对之间的相关系数。
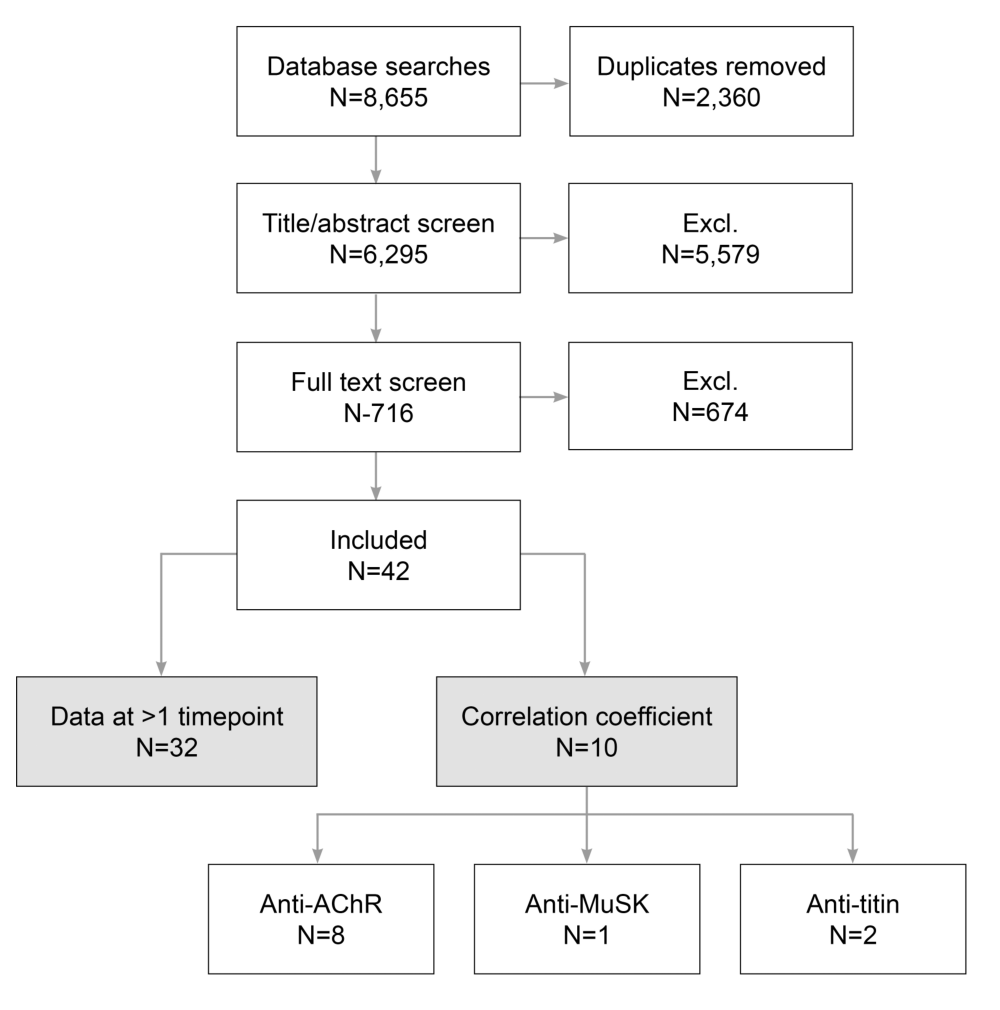
表2 Evidence summary by autoantibody, study design, and outcome (n = 10)
方法:2020年10月,来自欧洲各地的神经学领先临床医生和研究人员的论坛(重症肌无力神经学罕见自身抗体专家论坛)参加了一系列虚拟会议,同时进行了系统的文献回顾(SLR)。
结果:42项研究符合纳入标准。在这些患者中,有10人报告患者的自身抗体水平与疾病严重程度之间存在一定的相关性。一般来说,自身抗体水平的降低(乙酰胆碱受体、肌肉特异性激酶和肌蛋白)与疾病严重程度(重症肌无力定量评分、重症肌无力综合评分、重症肌无力日常生活能力评分、美国重症肌无力基金会分级)的改善呈显著正相关。鉴于证据有限,测试预定义变量的影响是不可行的。
结论:首次评估MG患者自身抗体水平与疾病活动性之间是否存在相关性的SLR显示了潜在的正相关性,这可能对指导治疗决策具有临床意义。然而,考虑到有限和可变的证据,我们目前不建议在这方面进行自身抗体水平检测的常规临床应用。目前,患者的特征、临床病程和实验室数据(例如,自身抗体状态、胸腺组织学)应该与患者报告的结果一起为治疗提供信息。我们强调,未来的研究需要就这种关系得出更明确的结论。
原始出处:
作者:医路坦克
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







在大多数患者(约85%)中,致病自身抗体针对#烟碱型乙酰胆碱受体#(AChRs),其他自身抗体针对#肌肉特异性激酶#(约6%的患者)和#低密度脂蛋白受体相关蛋白4#(LRP4;约2%的患者)。 一般来说,#自身抗体#水平的降低(#乙酰胆碱受体#、#肌肉特异性激酶#和#肌蛋白#)与疾病严重程度(重症肌无力定量评分、#重症肌无力#综合评分、重症肌无力#日常生活能力评分#、美国重症肌无力基金会分级)的改善呈显著正相关。
62