肌萎缩侧索硬化症:症状与体征、病因、流行病学、诊断和治疗
2022-08-29 MedSci原创 MedSci原创
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS),也叫运动神经元病(MND),后一名称英国常用,法国又叫夏科(Charcot)病,而美国也称卢伽雷(Lou Gehr
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS),也叫运动神经元病(MND),后一名称英国常用,法国又叫夏科(Charcot)病,而美国也称卢伽雷(Lou Gehrig)病, 是一种病因未明、主要累及大脑皮质、脑干和脊髓运动神经元的神经系统变性疾病。其局限性分型包括进行性球麻痹(PBP),连枷臂、腿,进行性肌萎缩(PMA),原发性侧索硬化(PLS)。我国通常将肌萎缩侧索硬化和运动神经元病混用。ALS 以进行性发展的骨骼肌萎缩、无力、肌束颤动、延髓麻痹和锥体束征为主要临床表现。一般中老年发病,生存期通常 3~5 年。
一、一般概述
肌萎缩侧索硬化症 (ALS) 是一种神经退行性疾病,其特征是大脑、脑干和脊髓中的神经细胞(神经元)进行性退化并最终死亡。 ALS 中涉及的神经元促进神经系统和身体随意肌(运动神经元)之间的交流。通常,大脑中的运动神经元(上运动神经元)向脊髓和脑干中的运动神经元(下运动神经元)发送信息,然后将信息传递给各种肌肉。 ALS 会影响上运动神经元和下运动神经元,因此信息的传递会中断,肌肉会逐渐变弱并消耗殆尽。结果,失去了启动和控制自主运动的能力。 ALS 会影响移动手臂和腿、说话和吞咽、支撑颈部和躯干以及呼吸所需的肌肉。 ALS 的症状会随着时间的推移而发展,最终导致呼吸衰竭,因为受影响的个体失去了控制胸部和横膈膜肌肉的能力。尽管在美国有两种疗法被批准以少量(疾病修饰疗法)减缓疾病的进展,但 ALS 的主要治疗方法集中在症状控制和支持性护理上。
二、症状与体征
ALS 会导致上运动神经元和下运动神经元疾病的组合,并且症状会因受累神经元控制的肌肉以及上运动神经元或下运动神经元受到主要影响而有所不同。上运动神经元病的主要表现是肌肉无力、肌张力和僵硬增加(痉挛)、反射增加(反射亢进)以及言语和吞咽异常。下运动神经元疾病会导致肌肉无力和消瘦(萎缩)、肌张力下降、反射减弱(反射减退)、肌肉纤维抽搐(肌束震颤)、肌肉痉挛以及言语、吞咽和呼吸异常。当 ALS 影响四肢和躯干肌肉时,会导致行走或跌倒困难以及日常生活活动困难等症状。当 ALS 影响头颈部神经(颅神经)时,它会导致延髓症状,包括吞咽困难(吞咽困难)或说话困难(构音障碍)以及面部或舌头肌肉无力。吞咽困难会导致并发症,例如进食困难、窒息、唾液过多或流口水,以及体重减轻。当食物或液体由于吞咽功能障碍进入气道时,吞咽困难也可因吸入食物内容物(吸入性肺炎)而导致肺炎。延髓症状还可能包括以突然、无法控制和不恰当的笑或哭(假性延髓影响)为特征的情绪不稳定。
尽管 ALS 的症状可以在成年期的任何时候开始,但它们最常见于 55 至 75 岁的个体。儿童期发病的遗传性 ALS 非常罕见。在疾病早期,患者可出现上运动神经元症状和下运动神经元症状中的一种或两种。症状最常见于四肢(脊髓性 ALS)。四肢出现的症状会影响上肢和下肢中的一个或两个,并且最初通常在一侧(不对称)更明显。早期,ALS 的症状可能很微妙,包括轻微的肌肉无力、手部动作笨拙和/或难以执行需要手指和/或手部精细动作的任务。腿部肌肉无力可能导致绊倒和跌倒。大约三分之一的患者最初表现为主要的延髓症状(延髓性 ALS)。更罕见的是,患者最初可能会因呼吸肌无力而出现呼吸症状,例如呼吸急促(呼吸困难)。 ALS是一种神经退行性疾病,因此症状会随着时间的推移而恶化;肌肉受到更严重的影响,并涉及更多的肌肉。该疾病可能进展迅速或缓慢。随着 ALS 的进展,通常在三到五年的过程中,个体将逐渐失去站立或行走的能力。随着时间的推移,许多患者将需要机械辅助呼吸,并且呼吸衰竭的风险增加。一小部分 ALS 患者的症状会逐渐稳定,并且可能会在几个月或很少几年内维持该水平(平台期)。
尽管 ALS 主要被视为一种影响运动神经元的疾病,但在多达一半的受影响个体中可以观察到非运动症状。事实上,大约 10% 的患者同时受到一种称为行为变异性额颞叶痴呆 (bvFTD) 的疾病的影响,并可能出现认知障碍和行为症状,例如去抑制、暴饮暴食和强迫或不当行为(有关这种疾病的更多信息,请选择“额颞叶退化”作为您在罕见病数据库中的搜索词)。在 ALS 中可以看到的其他非运动症状包括认知障碍和行为变化,这些变化通常不如 FTD 患者明显,情绪改变如抑郁和假性延髓影响(如上所述)。此外,ALS 患者可能因活动能力下降而发生血栓的风险增加。
三、病因
散发性 ALS 的根本原因尚不清楚。人们认为,多种相互关联的分子机制的功能障碍会导致该疾病。这些机制包括蛋白质平衡、折叠和运输的功能障碍、过度的神经元刺激(兴奋性毒性)、氧化应激、神经炎症和线粒体(“细胞的发电站”)功能障碍。最终,这些异常会导致运动神经元的损伤和死亡,从而导致 ALS 的症状。只有年龄和家族史是明确确定的 ALS 风险因素。
大约 10% 的 ALS 病例是家族性的(遗传性的)。超过 25 个基因与这种疾病有关。大多数家族病例遵循显性遗传模式,尽管也可能存在隐性或 X 连锁遗传模式。然而,一些具有致病(致病性)基因突变的个体不会患上这种疾病(不完全外显率)。基于已知基因突变的存在,通常无法准确预测发病年龄和疾病特征。
家族性 ALS 最常由 C9ORF72 基因突变引起。这种突变可导致 ALS、额颞叶痴呆 (FTD) 或两者兼而有之。家族性 ALS 的第二个最常见原因是由于 SOD1 基因的突变。几乎一半的家族性 ALS 病例是由 SOD1 和 C9ORF72 基因突变引起的,另外 20% 是由称为 TARDBP 和 FUS 的基因突变引起的。这些基因的突变都与家族性 ALS 的显性形式有关。
当只需要一个异常基因的一个拷贝来引起特定疾病时,就会发生显性遗传疾病。异常基因可以遗传自父母中的任何一方,也可以是受影响个体中基因突变的结果。每次怀孕将异常基因从受影响的父母传给后代的风险为 50%。男性和女性的风险相同。
基因检测 自 20 世纪 90 年代以来,已发现 SOD1、 ANG 、 VAPB 、 VCP 、SQSTM1、 TARDBP 、 DCTN1、 DAO 、 SETX 、 FUS 、 C9ORF72、 ATXN2 、 OPTN 、 SCFD1 、 NEK1 、 C21ORF2 等 20 多个基因突变。建议充分、详细询问 ALS 患者及其兄弟姐妹的病史以及患者父母、祖父母的详细病史和其兄弟姐妹的病史。
四、流行病学
ALS 是一种罕见的疾病,在北美和欧洲人口中每年每 100,000 人中有 1.5 至 3 人发病。 美国约有 30,000 人受到影响,估计每年诊断出 5,000 例新病例。 ALS 影响的男性多于女性,因为大约 60% 的受影响个体是男性。 需要对 ALS 的流行病学进行进一步研究,因为绝大多数流行病学研究都集中在北美和欧洲人群。
欧洲及美国年发病率是 2/10 万 ~3/10 万,患病率为(3~5)/10 万。发病的高峰年龄为50~75 岁,不随着年龄增加而增高。约 10% ALS 患者为家族性,余 90%为散发性。ALS 中男女患病率比例为(1.2~1.5):1。家族性ALS 的平均发病年龄较散发性ALS 发病年龄早。中国 ALS 的流行病学数据主要来自中国(中国香港地区),发病率约 0.6/10 万人,患病率约 3.1/10 万人。
五、鉴别诊断
ALS 变体
有几种运动神经元疾病亚型与 ALS 有一些重叠的特征和病理学。与经典 ALS 相比,这些运动神经元疾病表型通常与更好的预后相关。在生物学上,尚不清楚这些实体是否与 ALS 连续存在,或者它们是否代表明显不同的疾病。
原发性侧索硬化症 (PLS) 是一种罕见的神经系统疾病,其特征是上运动神经元进行性丧失。上运动神经元丢失会导致手臂和腿部肌肉痉挛和无力,表现为行走不稳、绊倒或使用手或手臂困难。延髓肌肉中的上运动神经元缺失会导致言语和吞咽异常,尽管延髓症状通常不是 PLS 的最初表现症状。 PLS 会导致进行性残疾,但通常进展比 ALS 慢得多。与 ALS 不同,PLS 不影响下运动神经元。 (有关这种疾病的更多信息,请在罕见病数据库中选择“原发性侧索硬化症”作为您的搜索词。)
进行性肌肉萎缩 (PMA) 仅涉及下运动神经元的进行性丧失。这种疾病的特点是肌肉无力和萎缩,尤其是腿部。检查时注意到语气和反射减弱。如果上运动神经元症状在两年内没有出现,那么将来发生 ALS 的可能性较小。
局灶性或单体性肌萎缩仅影响身体的一个区域的下运动神经元,最常见于手和手臂的肌肉。受影响的肌肉会出现萎缩和虚弱。发病通常发生在成年早期。该疾病通常会在几个月内进展,然后使患者出现固定的功能障碍。
其他相关疾病
以下疾病的症状有时可能具有与 ALS 相似的特征。比较可能有助于鉴别诊断。
脊髓性肌萎缩症 (SMA) 是一组罕见的遗传性神经系统疾病,最常见于婴儿期,其特征是下运动神经元的进行性退化。 SMA 最常见于婴儿期或儿童期,但对于主要表现为四肢下运动神经元疾病的患者,在鉴别诊断中可考虑成人发病的 SMA。 (有关这种疾病的更多信息,请选择“脊髓性肌萎缩症”作为您在罕见病数据库中的搜索。)
多灶性运动神经病 (MMN) 是一种罕见的疾病,其特征是下运动神经元功能障碍,主要是手臂和腿部。这种疾病被认为是免疫介导的,这意味着由于免疫系统功能异常和存在针对体内特定蛋白质的特定自身抗体而导致炎症。术语多焦点的意思是“来自两个或多个解剖位置”。这种疾病是在一个人一生中的某个时刻获得的;一个人并非天生就有这种疾病。多灶性运动神经病通常对静脉注射免疫球蛋白 (IVIG) 治疗有反应。 MMN 被认为是肢体无力患者的鉴别诊断,尤其是手部无力,并且检查结果与下运动神经元功能障碍一致。 (有关这种疾病的更多信息,请选择“多灶性运动神经病”作为您在罕见疾病数据库中的搜索。)
遗传性痉挛性截瘫 (HSP) 是一大类遗传性疾病,主要影响上运动神经元。 HSP 的主要症状是由于腿部肌肉无力和痉挛而导致行走困难。 HSP有80多种不同的遗传类型。腿部无力的严重程度(从无到显着)、痉挛程度(从最小到严重)和其他神经系统症状的发生在不同遗传类型的 HSP 之间可能存在显着差异,以及性质差异具有完全相同的 HSP 基因类型的个体之间的症状严重程度。对于以上运动神经元症状为主的患者,尤其是腿部症状,在鉴别诊断中考虑 HSP。 (有关这种疾病的更多信息,请选择“遗传性痉挛性截瘫”作为您在罕见病数据库中的搜索。)
肯尼迪病或脊髓延髓肌萎缩症是一种罕见的 X 连锁疾病,可导致成年男性进行性下运动神经元疾病和雄激素功能障碍症状。
六、诊断
ALS是一种临床诊断。这意味着没有单一的测试可以可靠地诊断出这种疾病。因此,ALS 的诊断集中在仔细的患者病史和神经系统检查上。根据临床表现,实验室和影像学检查可能有助于排除其他疾病。 ALS 的诊断需要有扩散到一个或多个解剖区域的进行性肌无力病史和上、下运动神经元疾病的临床证据,尽管在病程早期可能只有一种运动神经元功能障碍占主导地位(见体征和上面的症状部分详细了解上下运动神经元病的临床表现)。电诊断研究,如肌电图 (EMG) 和神经传导研究 (NCS),评估神经冲动向肌肉的传递和神经冲动在神经元之间的传导,可以补充体格检查并显示运动神经元功能障碍的进一步证据。脑成像,例如磁共振成像 (MRI),通常在疑似 ALS 的患者中进行。虽然在 ALS 中可以看到某种程度的脑萎缩,但成像主要是为了排除运动神经元疾病的其他原因。基因检测对疑似家族性 ALS 的病例特别有用。诊断延迟是 ALS 的常见问题,从症状出现平均诊断延迟 1 年。
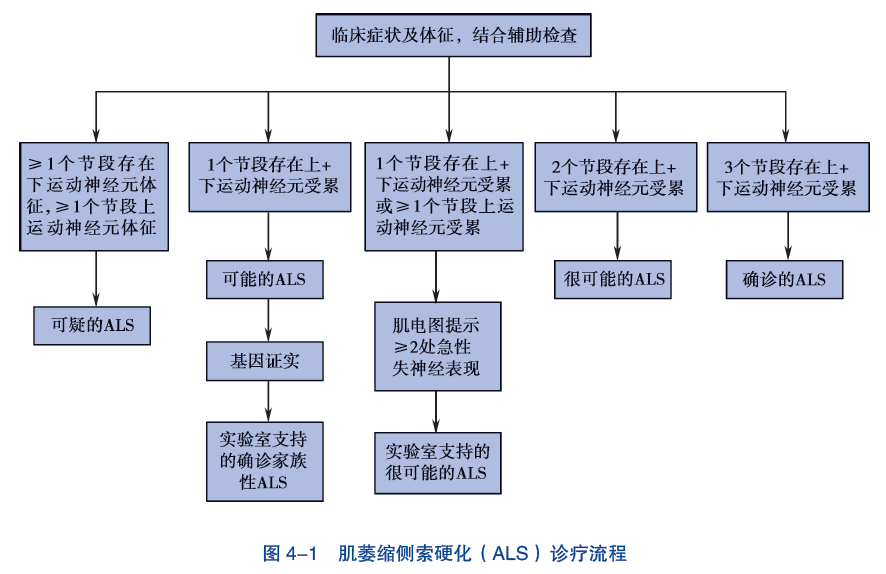
ALS 的早期临床表现多样,缺乏特异的生物学确诊指标。详细的病史、细致的查体和规范的肌电图检查对于早期诊断具有关键性的作用。影像学等其他辅助检查在鉴别诊断中具有一定价值。根据患者所出现症状、体征的解剖部位,可分为脑干、颈、胸和腰骶 4 个区域;根据临床和肌电图检查所证实的上、下运动神经元受累区域多少,可分为不同的ALS 诊断级别(EI Escorial 标准修订版)(表 4-1)。
1. 临床确诊 ALS 通过临床或电生理检查,证实在 4 个区域中至少有 3 个区域同时存在上、下运动神经元同时受累的证据。
2. 临床拟诊 ALS 通过临床或电生理检查,证实在 4 个区域中至少有 2 个区域同时存在上、下运动神经元同时受累的证据。
3. 实验室支持的拟诊 ALS1 个区域上、下运动神经元同时受累或仅有上运动神经元受累伴电生理检查提示至少 2 个区域的下运动神经元受累,影像和实验室检查排除其他疾病。
4. 临床可能 ALS 通过临床或电生理检查,证实仅有 1 个区域存在上、下运动神经元同时受累的证据,或者在 2 个或以上区域仅有上运动神经元受累的证据。已经行影像学和实验室检查排除了其他疾病。

诊疗流程
七、治疗
ALS 的治疗通常需要多学科团队合作,尤其应包括神经科医生、物理治疗师、言语病理学家、肺科医生、肺治疗师、医疗社会工作者、营养师、心理学家和专业护士。 ALS 的多学科护理与提高生存率和患者满意度有关。
ALS 治疗有两个主要组成部分:减缓疾病进展的疗法(疾病修饰疗法)和有助于控制症状和改善生活质量的疗法(支持疗法)。不幸的是,没有治愈 ALS 的方法。
疾病改善疗法
利鲁唑 (Rilutek) 是美国食品和药物管理局 (FDA) 批准用于治疗 ALS 的第一个药物。在临床试验中,利鲁唑被证明可以将生存期平均延长三到五个月,尽管它并没有显着延缓肌肉退化。另一种 FDA 批准的 ALS 疾病缓解疗法是依达拉奉(Radicava)。它被证明可以减缓某些 ALS 患者的功能衰退速度。早期 ALS 患者的益处似乎更为明显。
对症治疗
ALS 的对症治疗有两个主要组成部分:药物治疗和非药物治疗。
可以使用几种药物来帮助缓解 ALS 的症状。肌肉痉挛和肌束震颤可以用肌肉松弛剂如巴氯芬、替扎尼定或地西泮来控制。在一些患有严重和致残痉挛的患者中,巴氯芬可能会使用称为鞘内泵的装置直接注入椎管(鞘内给药)。一些患有痉挛的人也可能从大麻素治疗中受益。肌肉痉挛可能很痛,可以用硫酸奎宁、左乙拉西坦或美西律等药物治疗。一些 ALS 患者可能会出现唾液分泌过多(流涎)并且无法控制分泌物的汇集;这可以通过阿托品、东莨菪碱、阿米替林、格隆溴铵或肉毒杆菌毒素注射等药物来控制。口腔吸引装置也有好处。情绪改变,如抑郁症或与额颞叶痴呆相关的行为症状可以用抗抑郁药如选择性5-羟色胺再摄取抑制剂(SSRIs)来控制。一些 ALS 患者可能会因不同原因而感到疼痛,可以根据疼痛的类型使用多种药物进行治疗。
物理和职业治疗非常重要,应该包括每天的活动范围练习。这些练习可以帮助保持受影响关节的灵活性并防止肌肉固定(挛缩)。 ALS 患者保持适当的营养也很重要。体重减轻是 ALS 预后不良的独立预测因素。吞咽困难患者应慎重选择软食。当因吞咽困难而无法维持足够的营养和液体时,可以考虑使用胃饲管。言语治疗和增强通信设备对构音障碍患者很有用。
一旦个体出现呼吸肌无力,无创正压通气 (NIPPV) 有助于辅助呼吸。咳嗽辅助装置也可用于清除分泌物。最终,通气无力进展到患者无法自主呼吸,一些患者会选择进行气管切开术和永久机械通气。对于决定不使用机械通气的患者,家庭临终关怀服务可以提供支持性护理并协助采取舒适措施。
八、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Schultz J. Disease-modifying treatment of amyotrophic lateral sclerosis. Am J Manag Care 2018;24:S327-S35.
Hardiman O, Al-Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers 2017;3:17085.
Brown RH,Al-Chalabi A.Amyotrophic Lateral Sclerosis.N Engl J Med,2017,377(2):162-172.
Brooks BR,Miller RG,Swash M,et al.For the World Federation of Neurology Research Committee on Motor Neuron Disease.EI Escorial revisited:revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis,2000,1 :293-300.
Ludolph A,Drory V,Hardiman O,et al.Arevision of the EI Escorial criteria-2015.Amyotroph Lateral Scler Frontotemporal Degener,2015,16(5-6):291-292.
Elman LB, McCluskey L. Clinical features of amyotrophic lateral sclerosis and other forms of motor neuron disease, UpToDate. Last updated: Feb 11, 2021. https://www.uptodate.com/contents/clinical-features-of-amyotrophic-lateral-sclerosis-and-other-forms-of-motor-neuron-disease Accessed August 5, 2021.
Elman LB, McCluskey L. Diagnosis of amyotrophic lateral sclerosis and other forms of motor neuron disease, UpToDate. Last updated: Jun 02, 2021. https://www.uptodate.com/contents/diagnosis-of-amyotrophic-lateral-sclerosis-and-other-forms-of-motor-neuron-disease Accessed August 5, 2021.
Galvez-Jimenez N. Symptom-based management of amyotrophic lateral sclerosis. Last updated: Apr 20, 2021. https://www.uptodate.com/contents/symptom-based-management-of-amyotrophic-lateral-sclerosis Accessed August 5, 2021.
McCluskey L. Familial amyotrophic lateral sclerosis, UpToDate. Last updated: Mar 23, 2021. https://www.uptodate.com/contents/familial-amyotrophic-lateral-sclerosis Accessed August 5, 2021.
Galvez-Jimenez N, Goyal NA, Cudkowicz ME. Disease-modifying treatment of amyotrophic lateral sclerosis, UpToDate. Last updated: Nov 09, 2020. https://www.uptodate.com/contents/disease-modifying-treatment-of-amyotrophic-lateral-sclerosis Accessed August 5, 2021.
Maragakis NJ, Galvez-Jimenez N. Epidemiology and pathogenesis of amyotrophic lateral sclerosis, UpToDate. Last updated: Apr 20, 2021. https://www.uptodate.com/contents/epidemiology-and-pathogenesis-of-amyotrophic-lateral-sclerosis Accessed August 5, 2021.
Siddique N, Siddique T. Amyotrophic Lateral Sclerosis Overview. 2001 Mar 23 [Updated 2019 Oct 3]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1450/ Accessed August 5, 2021.
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#萎缩#
240
#硬化症#
161
学习了
127
#流行病#
94
#肌萎缩#
108