
NEJM综述:基因组与癌症治疗
2011-04-05 MedSci原创 MedSci原创
MedSci评:这是大师级的综述,值得反复阅读学习。下面翻译来自医学论坛网。有些翻译不是很规范,如果需要的话,可以阅读英文全文。 2000年1人类基因组序列的提供激起了几轮癌症研究的热潮。一套基本上完整的蛋白编码基因的识别,加上新型转录元件如微小RNA(MicroRNA)的发现,已经助长了一次研究的暴发,这些研究采用基于阵列(Microarray)的方法,对大多数癌症类型中的基因表达模式谱进
MedSci评:这是大师级的综述,值得反复阅读学习。下面翻译来自医学论坛网。有些翻译不是很规范,如果需要的话,可以阅读英文全文。
2000年1人类基因组序列的提供激起了几轮癌症研究的热潮。一套基本上完整的蛋白编码基因的识别,加上新型转录元件如微小RNA(MicroRNA)的发现,已经助长了一次研究的暴发,这些研究采用基于阵列(Microarray)的方法,对大多数癌症类型中的基因表达模式谱进行研究。同样,识别体细胞突变的系统方法的研发,促进了对癌基因组变化的彻底分析,包括拷贝数变化(DNA的缺失和扩增)、重排、小的插入和缺失,以及点突变2。最近,这些努力已在以下两个方面达到高潮:对人类癌症的全基因组进行测序,以及提供体细胞突变的综合目录3,4。这些研究已深入阐明了可促成细胞转化的基因2。同时,人群中遗传变异的特征描述已发动了一波探索癌症易感性的浪潮,主要集中在常见于一般人群以及使癌症危险小幅增加的DNA变异体。最后,研究者已开发出数种成套的生物试剂,它们可干扰活细胞中基本上所有基因的功能,最广泛使用的是小干扰RNA。这些试剂以形形色色的方式被使用——例如,系统地确定哪种基因是癌细胞生存所需要的,以及哪种基因可赋予(癌细胞)对特定药物的敏感性。
这种研究的一些早期成果,连同应用这些成果所需要的技术,已被合并为临床肿瘤学。在本文中,我们将就以下内容进行综述:基因组方法对肿瘤分类、预后标志物、药物疗效反应的预测指标、新药物疗法的研发、监测疾病的策略以及对癌症易感性的处理产生的影响。
生物学分类
对于大多数癌症,我们仍然依赖于染色的组织切片或细胞的组织学分析进行诊断和亚分类。在某些肿瘤类型(例如乳腺癌和白血病)中,分子标志物已成为组织学分类的辅助物达数十年。以微阵列为基础的(基因表达)谱[在单次实验中可检测数以千计信使RNA(mRNA)转录物的表达水平]的问世,已明显提高了对癌症进行子分类的能力。最著名的例子可能是(基因)表达谱对乳腺肿瘤分类的作用5。虽然病理科医师很久以前就已经知道该病的异质性,但(基因)表达谱已促使(人们)开发出一种通常包括下述重要分子亚型的分类:基底(细胞)样型、人类表皮生长因子受体2(HER2)阳性型、正常乳腺样型、管腔 A型和管腔B 型。这种分类仍在不断进展,但已成为临床乳腺肿瘤学中广泛使用的一种概念框架,即不同亚型具有明显不同的临床和生物学特征,包括病人的生存情况6,7。然而,在日常的临床实践中,仍基于常规的组织学分析,结合雌激素受体(ER)、孕激素受体(PR)和HER2的免疫组化染色进行分类,当这些检查结合在一起时,可重建用mRNA表达定义的大多数子类。
除了细化肿瘤分类外,这些进展可能被如何应用呢?一个常规组织学分析经常力求解决的一个标准临床分类问题的范例是,确认转移癌的原发组织来源。然而,基因表达标签可提供超过在显微镜辅助下人眼的分辨敏锐度,并且由于癌中经常保留了来源组织表达模式的元件,因此对(基因)表达谱(特别是那些包含表达的微小RNA者)的分析,常可提供额外的信息,尽管目前它们尚未被用于日常的临床实践8。
预后指标
基因表达标签(EST)的使用已经使(人们)能够识别出预后的子类。例如,在有早期乳腺癌的病人中,基因表达谱已被用来定义复发的危险。三种(表达)谱已经显示具有预测能力:70-基因谱(MammaPrint),已获得美国食品和药物管理局(FDA)的批准;21-基因复发评分(Oncotype DX);以及76-基因转归鹿特丹标签9-11。这些(表达)谱通过使用肿瘤源性mRNA获得,后者是根据DNA微阵列或定量逆转录—聚合酶链反应(RT-PCR)分析得到测定的,目前在临床实践中这些(表达)谱被用来预测(病人)预后,并因此影响治疗干预。
Oncotype DX测定法的使用已被纳入2007年美国临床肿瘤学会的指南中,用于评估有淋巴结—阴性、ER-阳性疾病的病人,以便识别出有可能从他莫昔芬辅助治疗中获得最大益处的病人,以及不一定需要化疗的病人。引人注目的是,最近一项涉及医师实践的多中心研究显示,根据这种测定法,内科肿瘤医师已改变了对该亚组中几乎三分之一病人的治疗建议12。
以同样的方式,一种12-基因表达标签已被开发用于结肠癌。在有Ⅱ期肿瘤的病人中,它是复发的一个独立预测因子,并可能有助于更准确地定义不会从化疗中获益的病人13。在血液系统癌症中,基因表达谱在有正常核型急性髓系白血病成年病人中识别出了一种133-基因标签,后者可提供独立于其他已知不良特征的预后信息14。与此相似,基因表达分析已经在弥漫性大B细胞淋巴瘤(DLBCL)中识别出新的预后亚型,这可改善伯基特(Burkitt)淋巴瘤与 DLBCL的鉴别难度14,15。
然而,人们对在有合适(统计学)效力的随机、对照试验证实基因表达标签的有效性之前,将其并入常规临床实践中存在担忧。这种担心已促使(研究者开展了)以下大型试验,以检验MammaPrint和Oncotype DX在淋巴结—阴性乳腺癌病人中预测辅助化疗益处方面的有效性:检验 MammaPrint的在淋巴结阴性和1~3个淋巴结阳性的疾病中微阵列有可能避免化疗试验(MINDACT)(ClinicalTrials.gov,编号NCT00433589),以及检验Oncotype DX的给予个体化治疗选择试验(TAILORx)(NCT00310180)(图1)。虽然两项研究在很大程度上都将有助于在早期乳腺癌治疗中(制定)循证决策,但值得注意的是,每项研究都需要纳入数以千计的病人,以遵循确切回答这一具体问题所需的复杂试验设计。很关键的一点是,任何新的试验都应在病人中经受任何新药或操作所需的同样严格的评价,因此复发危险的新标签(如结肠癌中所报告的)也应在前瞻性、随机临床试验中得到检验16。虽然这些类型的临床试验需要的样本量大,但技术进步(例如使基因-表达标签能够从福尔马林固定组织中获得的技术) 
优化治疗学的使用
执业肿瘤科医师的共有经历是,患特殊类型癌症的病人仅有一个亚组将从特定疗法中明显获益。鉴于肿瘤之间存在极大的基因组学异质性,所以病人对相同治疗存在疗效反应上的差异并不令人惊讶。部分这些基因组学差异在确定临床治疗反应的可能性方面有重要作用。特别是,在治疗药物靶向作用于特定细胞蛋白的情况下,编码那种蛋白基因的基因组改变可能是疗效反应的重要决定因素。例如,乳腺癌中HER2的超表达和扩增是(病人)可以从曲妥珠单抗(一种针对HER2的抗体)治疗中获益的一个强预测因子17。根据这种研究结果, 1998年FDA批准曲妥珠单抗用于治疗HER2扩增的转移性乳腺癌。
继这一例子和其他例子之后,已有人推测,在过去的10年里,大多数进入临床试验的新药可影响对癌细胞增殖或生存重要的通路。参与这些细胞通路的基因时常由于体细胞改变(点突变、缺失、扩增和易位)而发生突变,被称为驱动突变,这可直接促成癌细胞的异常生长。因此,这些基因存在突变与否,将对一例病人对特殊靶向疗法的疗效反应产生深远影响。
例如,表皮生长因子受体(EGFR)激酶活性的小分子抑制剂最初被开发用于癌症治疗,因为EGFR在调节细胞增殖方面的已知作用,以及因为该基因在许多癌中超表达。后来,有人发现在非小细胞肺癌中有EGFR的活化体细胞突变,这一发现确定了一个对EGFR抑制剂有特别好的疗效反应的病人亚组 18,19。一项大型前瞻性研究目前已经显示,在肿瘤有一种活化EGFR突变的非小细胞肺癌病人中,靶向EGFR抑制剂的有效率为71%,相比之下,没有突变者的有效率是1%20。由于该试验和其他相似试验的结果,研究者对肿瘤活检样本进行了分析,目的是确定可赋予靶向药物敏感性的癌基因中关键突变的一个亚组,在某些医学中心,该分析已作为常规诊断性检测被引进。
在某些情况下,一种治疗药物有可能与一个突变基因具有协同作用,即使该药不直接靶向作用于突变基因编码的蛋白上也如此。例如,多[腺苷二磷酸(ADP)-核糖]聚合酶(PARP)蛋白抑制剂(抑制一种类型的DNA修复),在有双链断裂修复缺陷(由于BRCA1或BRCA2失活突变)的癌中似乎特别有效21。这种协同作用,经常被称为协同致死性22,被认为其发生是肿瘤细胞中未修复的DNA损伤负荷过大(使两种DNA修复过程均出现缺陷)的结果。
有人预期,随着表明特殊基因改变可预测疗效反应证据的出现,当前在研发流水线中的许多新治疗药物将进入医院(表1)。如果这种情况的确发生,我们将需要对每种癌症进行一连串不断增加的诊断性基因检测,以便用最有效和最节俭的方式来配置这些药物。最近对实体瘤进行的测序研究已显示,许多癌基因在不到 10%的任何单一类型的肿瘤中发生突变。因此,如果这种靶向疗法普遍有效,将需要一份大得多的药物清单(以及诊断性检测),以优化治疗大多数有一种特定类型癌症的病人23。 
新疗法的研发
研发靶向作用于由突变癌基因编码蛋白的抑制剂,目前已不断获得成功。在慢性髓系白血病(CML)中,最好用伊马替尼(格列卫)来举例说明,该药是一种埃布尔森(ABL)激酶的有效抑制剂。1960年在大多数有CML的病人中发现了费城染色体,这为最终使该病的治疗发生革命性变化的一系列发现创造了条件 24。该基因组学改变导致位于染色体22(BCR所在位置)与染色体9(ABL所在位置)长臂上的2个基因发生相互易位,从而导致一种嵌合型致癌融合蛋白(BCR-ABL)的形成,在此过程中ABL激酶被活化。伊马替尼治疗CML的临床试验显示,它明显优于常规化疗,在美国, FDA于2001年批准伊马替尼作为CML的一线治疗25。2006年的一项研究显示,新近被诊断为慢性期CML并接受伊马替尼治疗的病人,其5年总生存率为89%26。
伊马替尼在CML中获得的成功已被迅速扩展到胃肠间质瘤(GIST),大多数这种肿瘤可使一种被突变活化的受体酪氨酸激酶(称为c-Kit)产生突变 27。因为伊马替尼可有效抑制c-Kit,因此其对GIST的治疗作用在临床试验中得到了检验,并获得了>50%的客观有效率28,与常规化疗<5%的有效率相比具有优势29。
虽然这些都是将癌症特有基因组学改变的产物作为治疗靶点的重要例子,但涉及的突变基因均在郑重其事地开始参照人类基因组测序之前被发现。然而,在过去的几年里,对癌基因组进行的越来越广泛的系统测序,已识别出若干种新的突变癌基因。其中一些突变癌基因,特别是编码酶(如激酶)的癌基因,使它们编码的蛋白作为新的治疗靶点变得可取,在这种情况下,突变可能导致组成性激活。例子包括BRAF、PIK3CA、FGFR2、JAK2、AKT1和 IDH119,30-34,并且针对由许多这些突变基因所编码蛋白的抑制剂的搜寻工作正在进行中。
一个值得注意的例子是,通过癌中体细胞突变而活化的一种蛋白质是BRAF(一种丝氨酸—苏氨酸激酶)。研究者通过在一组各种癌中系统地寻找候选癌基因,而识别出了BRAF的活化突变。随后该研究显示,BRAF在大约三分之二的恶性黑色素瘤中发生了突变34。由于转移性恶性黑色素瘤的治疗方法有限,因此BRAF突变的发现触发了学术和制药部门中的多项药物发现项目。涌现自这些项目的药物Ⅰ期临床试验的初步结果已经显示了非凡的活性35,而进一步的试验还正在进行中(图2)。从黑色素瘤中发现突变BRAF到临床试验显示有前景的靶向治疗,进展的时间不到10年,说明基因组学的药物研发与生物学相结合可提供惊人的发展步幅。
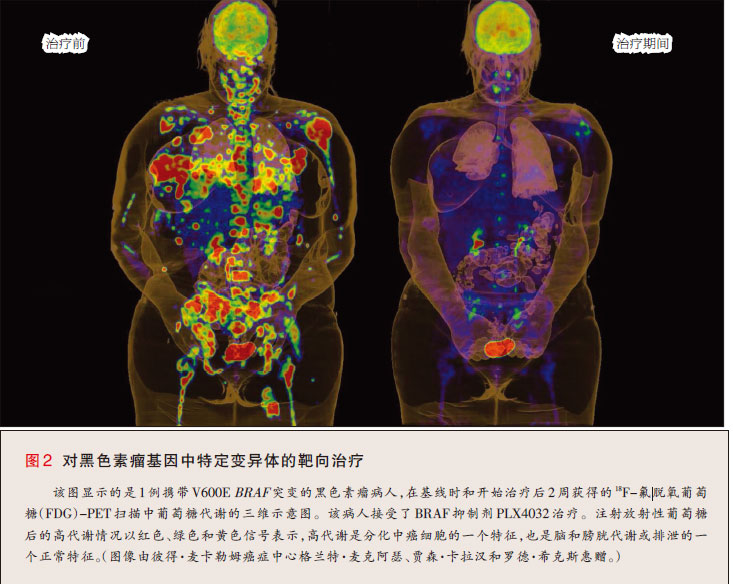
不幸的是,许多突变的癌基因并未产生针对新药研发易于驾驭的靶点。在这种情况下,研发显示与突变癌基因有协同致死性的药物似乎是一种明智的方法。例如,在 10%~ 20%的人类癌症中有RAS基因家族成员的突变和活化。研发可直接抑制致癌RAS药物的努力尚未成功。然而,小干扰RNA库的引入(每种小干扰 RNA在体外敲减癌细胞中数千个基因中一种基因的表达)已识别出突变KRAS与一对蛋白激酶(STK33和TBK1)之间有相互作用36,37。某些有 KRAS突变的细胞要依赖STK33和TBK1的表达才能生存,而有正常版本KRAS的细胞却不是这样。由于与KRAS本身相比,这些激酶有可能是更易于驾驭的药物靶点,因此STK33和TBK1抑制剂的研发阐明了一种潜在的治疗策略,该策略有可能应用于广谱的肿瘤。
大规模的癌症基因组研究,如国际癌症基因组联盟和癌症基因组图谱,已经对来自50种不同癌症类型的肿瘤应用了新一代测序技术,以便在基因组学、表观基因组学和转录物组学水平上生成25000多种癌基因组,并且将产生一个致癌突变的完整目录,其中某些突变有可能证明是新的治疗靶点38。有趣的是,最近两项研究表明,基因组随癌症的进展而演变。2个原发性乳腺癌以及后来转移灶的深度测序显示有新的突变,或者在转移癌中富集低频突变,这表明有以下可能性:对某些癌症转移(灶)组织进行的分析,或许能够识别出可成为重要药物作用靶点的额外突变39,40。
获得性治疗耐受
合理使用靶向突变基因产物的治疗已经获得了相当大的成功,但是许多病人因为其肿瘤变得对药物耐受而出现复发41。最初存在于癌细胞小部分亚克隆中的基因组改变,经常成为这些环境下获得耐药的基础。这些改变的范围从编码药物靶向蛋白基因内的一个附加点突变——如CML中的ABL突变和非小细胞肺癌中的 EGFR突变42-45 ——到完全不同癌基因的扩增,如非小细胞肺癌中的MET 46。
耐药机制的性质决定了二线治疗中所采用的治疗方法。这些方法有可能包括使用能克服靶基因中耐药突变的第二代抑制剂,与CML和非小细胞肺癌中的情况一样,或涉及靶向原发性突变基因和附加突变基因蛋白质产物的联合策略。
如果在原发性肿瘤中低频存在耐药克隆,一个重要的问题是它们是否能在早期被检出,从而影响最初的治疗选择。癌基因组的深度测序能够检出少量的耐药细胞,并因此能够预告联合治疗策略,后者或许可使耐药克隆不断扩展至主宰肿瘤细胞群的机会减至最小。
对疾病负担和早期复发的监测
对于大多数实体瘤,疾病负担的监测可通过各种成像检查方法实现,在几种类型的癌症中可通过测定循环中的组织—特异性标志物(例如前列腺癌中的前列腺特异性抗原)来进行补充(检查)。然而,对于某些癌症,血液样本中肿瘤特异性基因异常的检测已被用于量化肿瘤负荷。该策略在血液系统癌症中尤其富有成效,在这类癌症中,常规上(制定)治疗决策的依据是某些基因改变的水平。这一成功依赖于在许多血液系统癌症中发现的重复性基因重排。由于存在设计出有针对性扩增异常连接基因组片段的PCR引物的可能性,因此对于在技术上简单、高度敏感并特异的测定法而言,这类重排是理想的底物。例如,在有慢性髓系白血病(CML)并在治疗后量化的BCR-ABL转录物水平下降到1/100的病人中,通常有持久的疗效反应47,而BCR-ABL转录物水平升高,常表明已出现对伊马替尼耐药的肿瘤克隆的生长加速,从而触发了向其他疗法的转换。
该策略尚未被常规用于大多数实体瘤的临床管理,主要原因是缺乏已知的重复性基因易位或重排。然而,由于许多实体瘤中的濒死细胞将裸DNA释放到循环中,因此理论上肿瘤负担可通过血液取样(与对血液系统肿瘤所进行的血液取样相似)进行监测。在有实体瘤病人的外周血中已检测到有低水平的肿瘤特异性点突变 48。这些突变已被用来检出结直肠癌的复发,并且也被用来检测非小细胞肺癌病人(接受EGFR激酶抑制剂治疗)中是否存在继发性耐药突变 49,50。该策略在临床实践中难以广泛执行,是因为检出携带一个点突变DNA片断的一个小亚群在技术上具有挑战性。然而,大多数实体瘤携带对于每例病人而言特有的基因组重排。实际上,最近的研究已显示,采用新一代测序(法)在原发癌中检测到的重排,可被用来检测循环中的肿瘤DNA,其水平与肿瘤负荷相关51,52。因此,有可能以一种与监测血液系统癌症相似的方式来监测几乎所有实体瘤病人的癌症负担,(图3,相互作用图以及本文的全文均可从NEJM.org获取)。
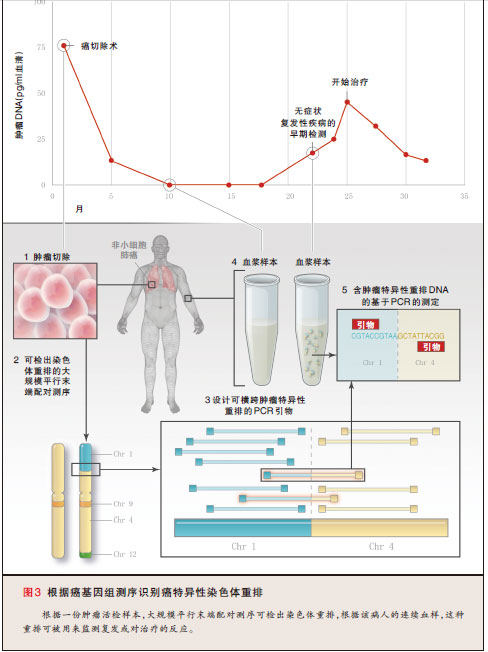
当前,大多数癌症早期复发的检出并未使(病人的)生存情况获得改善,并且在缺乏更有效疗法的情况下,这种情形将继续存在。我们希望这类疗法将从大多数主要癌症类型中的重复体细胞突变目录的形成中产生。然而,目前早期检出有可能对某些癌(例如结直肠癌)很重要,就这些癌而言,复发性疾病的手术切除可转化为生存情况的极大改善。该策略的实用性需要通过临床试验来严格评估。
临床试验设计中的基因组学
正如此前所述,许多新型抗癌药物的作用靶点是癌中有驱动性体细胞突变的基因所编码的蛋白。因此,许多这些药物有可能仅对肿瘤中存在这些突变的亚组病人有效。由于该亚组有可能相对(样本量)小,因此如果研究组由未经选择(根据突变状态)的病人组成,则某些药物的有效性不一定能被检出。该研究结果已促进了标准临床试验设计的改变,其中研究人群由具以下特征的病人充实起来:其肿瘤携带一种特定突变、成套突变或其他生物标志物,从而使这类肿瘤对干预措施敏感的可能性增加20,21,53。
然而,对于某些药物,目前还没有一种生物学决定子(是亚组病人反应性的基础)的先验指标(priori indication),因此不可能重新设置试验设计。尽管如此,目前未被发现的敏感性和耐药性的决定子,有可能被隐藏在每种肿瘤的基因组和转录物组中。随着基因组测序费用的急剧减少,人们可以在不远的将来,将癌基因组和转录物组作为癌症药物临床试验中的常规伴随物,考虑对它们进行完整的测序,以便揭示这些决定子。
对癌症的易感性
在2000年之前的数十年中,研究者已经发现了若干种遗传异常基因,它们使癌症的发生危险明显升高,并且存在于有多个受累成员的家族中。例子有 BRCA1和BRCA2(其中的突变使乳腺和卵巢癌的发生危险增高)以及MLH1和MSH2(其中的突变使结直肠癌的发生危险增高)。对这些在大多数人群中罕见的基因突变进行检测,目前是一种常规的临床方法。这些检测结果可影响筛查及其他的预防措施。
最近人类基因组序列的提供已允许对存在于正常人群的最常见遗传性DNA变异体进行编目54。这类工作已导致赋予癌症易感性的重要新一类变异体的识别。在大量患病的病人与对照受试者间,比较成百上千个遗传变异体患有率的全基因组关联性研究,已被应用于若干个癌症类型。这些研究导致了对赋予几个不同类型癌症易感性的许多新DNA变异体的识别,包括乳腺癌、前列腺癌、结肠癌和直肠癌、肺癌和睾丸癌,以及慢性淋巴细胞白血病和其他癌55。最初与一种肿瘤相关的遗传变异体不一定是它们本身的功能性改变,但它们通常位于基因组中紧邻真正致病性变异体的位置。
总体上,这些有可能存在于大多数人群中的常见易感性变异体,可使疾病的发生危险有极小幅的增加,从而使它们在临床实践中的实用性极为有限。然而,未来可能有机会在人群水平上一起检测若干种这类变异体,也许可识别出危险相对升高而进入疾病筛查项目的人群,或者有高危易感性突变引起的家族性疾病背景的人群,目的是进一步细化危险评估。
通过应用基因组技术和人类基因组序列所获得的发现,已经对肿瘤学实践的若干方面产生了影响,并且已经影响到临床试验的设计。在接下来的几年里,癌基因组研究的步伐将明显加快。协调全球倡议(项目)将产生成千上万种癌症的全基因组序列,从而得出每种癌症的体细胞突变的完整目录。这些研究将从本质上揭示突变癌基因的完整图谱,因此让我们更接近确立一个癌症发生的核心度量标准:多少突变癌基因以及什么样的突变癌基因组合是产生单个癌所必须的。这将澄清我们在癌症治疗中面临的难题。连同这些进展,对活癌细胞进行更系统的功能筛查来检出它们的生物学脆弱性,无疑将引导靶向药物研发的新方向。
今天作为研究工具的技术正准备成为明天诊断的手段。我们已经看到有限的RNA谱分析被引入到临床实践中,以及检测有限系列的体细胞基因异常以选择适合个体病人的靶向疗法。新一代测序技术的快速发展似乎可能是具有变革能力的。在几年内,花数百美元或更少钱就能获得完整的癌基因组序列。随着在各个癌中找到可提供信息的基因异常数量的持续增加,对全基因组进行测序最终有可能更节俭,而不是做一大组的直接检测。然而,为了开发癌基因组内信息在临床中的全部潜能,首先必须将对基因组和转录物组的分析更广泛地并入临床试验中,从而产生药物反应性和(病人)预后的预期之外的新预测因素。
原始文献:
Genomics and cancer care. Reference: N Engl J Med 2011; 364: 340-50.
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



#癌症治疗#
77