22岁女孩体内竟有上千个肿瘤,如野草般疯涨
2023-07-18 网络 网络
神经纤维瘤病(neurofibromatosis,NF)是一组常染色体显性遗传病,是基因缺陷使神经嵴细胞发育异常导致多系统损害,可以累及皮肤、外周或中枢神经系统、骨骼、脉管系统、内分泌系统等多个系统。
22 岁的丹丹在本该青春洋溢的年纪,却半瘫在床,而这一切的根源都是她体内密密麻麻的上千个如“泡泡”一样的肿瘤。在丹丹的口中,这是一种如同“疯长的野草”一样的病症。正是这种“疯长的野草”,让她的全身都被肿瘤占据。这种罕见病症是“神经纤维瘤病”。这种无孔不入的痛苦疾病,让全身凡是神经所能触及的地方,都将成为肿瘤生长的沃土。丹丹跨越 800 公里从河南来到北京看病。 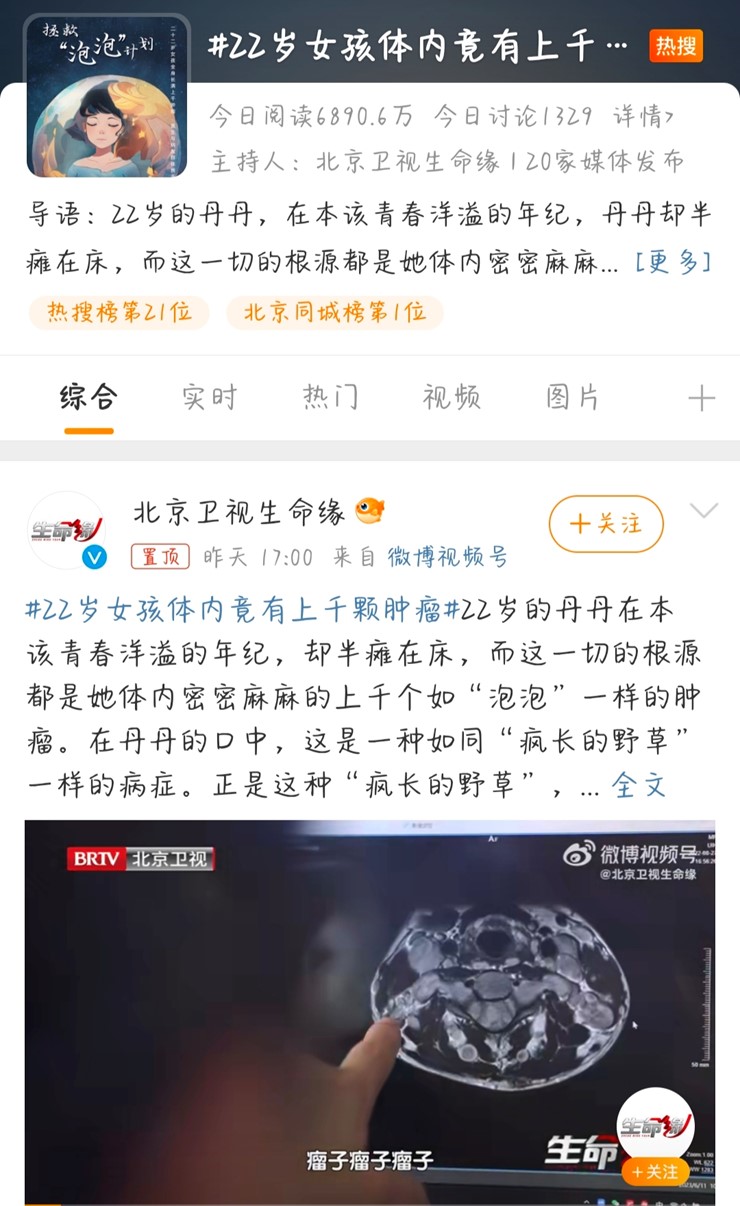
神经纤维瘤病(neurofibromatosis,NF)是一组常染色体显性遗传病,是基因缺陷使神经嵴细胞发育异常导致多系统损害,可以累及皮肤、外周或中枢神经系统、骨骼、脉管系统、内分泌系统等多个系统。
分类
神经纤维瘤病在临床上可分为3种主要类型:NF1、Ⅱ 型神经纤维瘤病(NF2)和神经鞘瘤病(schwannomatosis,SWN,又称施万细胞瘤病)。
- NF1:是神经纤维瘤病的主要类型,其患病率估测为 1/4000~1/2000 不等。主要表现为皮肤色素斑、骨骼损害(脊柱侧弯畸形)、癫痫、口腔损害、眼部病变(虹膜黑色素细胞错构瘤)、内分泌异常、内脏病变等等。
- NF2:发病年龄较 NF1 稍大,常发生在 20-40 岁。主要出现双侧听神经瘤,皮肤损害相对较轻。
临床上主要表现为双侧听力丧失、耳鸣。少数患者合并脑膜瘤、脊膜瘤、青少年后囊下晶状体混浊等。
神经鞘瘤病:
SWN 主要表现为中枢和外周混合性神经纤维瘤,没有虹膜色素瘤和视神经瘤。SWN 特征为多发性神经鞘瘤,常伴有剧烈疼痛。
病因
NF是由遗传或自身基因突变导致神经嵴细胞发育异常引起的。NF基因突变类型有错义变异、无义变异、剪切突变、小片段插入/缺失、大片段插入/缺失等多种形式。
Ⅰ 型神经纤维瘤病致病基因为位于染色体 17q11.2 的神经纤维瘤抑制基因(NF1),编码神经纤维瘤蛋白。大约 50% 的患者为新发变异,因此很多患者并没有家族史;而有家族史的遗传性致病性变异具有很高的外显率,但家族成员之间临床表现差异较大。
Ⅱ 型神经纤维瘤病致病基因为位于染色体 22q12.2 的 NF2 基因,编码神经膜蛋白,其失活与疾病密切相关。
神经鞘瘤病致病基因为位于染色体 22q11.23 的 SMARCB1 基因,编码的蛋白属于肿瘤抑制蛋白,参与基因的表观修饰和转录调控。此类型的患者大部分为新发变异,有家族史的患者中能检出SMARCB1 基因的患者仅占 40-50%。
临床表现
1、 NF1
(1)牛奶咖啡斑(CAMLs):
几乎所有的患者都有皮肤色素斑,呈淡棕色、暗褐色或咖啡色。腋窝部出现雀斑样色素沉着,这些斑点通常是永久性的,并可能随时间增大或数量的增加。生理变化如发育、妊娠、绝经、精神刺激均可使之加重,有时皮疹出现较迟,在发育期才开始发病,缓慢发展。
(2)多发性神经纤维瘤:
患者常诉全身出现无痛性皮下肿物,并逐渐增加和扩大。青春期和妊娠期明显进展。多无临床症状。少数表现为放射性或灼烧样疼痛,肿瘤压迫视神经引起视力下降等。
①皮肤型神经纤维瘤:cNF为最常见的神经纤维瘤类型。cNF常表现为橡胶状、外生软性丘疹和肿物,呈皮色或肉色,可发生在皮肤或皮下,大小不等,没有疼痛症状,多分布于面部、头皮、颈部、胸部。位于真皮层者,表面皮肤多为紫罗兰色变,边缘清楚,触之质软。较少见肿瘤呈软性皮下小肿块,略突出皮表,呈穹顶状外观,边界可触及,按压可平复(纽扣孔神经纤维瘤)。部分cNF会出现瘙痒或疼痛症状,与大量肥大细胞浸润相关。
②颅内肿瘤:可合并脑膜瘤,且可多发;合并胶质瘤、室管膜瘤,而出现癫痫、学习障碍等症状。
③椎管肿瘤:椎管内神经纤维瘤、脊膜瘤,可合并脊髓空洞或其它脊柱畸形。
④眼部症状:虹膜发生Lisch结节,实质为虹膜上的色素细胞错构瘤,裂隙灯下为橙黄色透明结节。
(3)神经症状:
部分患者可有轻度思维障碍、癫痫发作、肢体无力、麻木等。
(3)其它损害:
骨骼发育异常,如脊柱侧弯、颅骨缺损、长骨面骨过度生长、弯曲等。
2、 NF2
首发症状以双侧进行性听力下降最为常见,亦有部分病人表现为单侧严重的听力障碍或波动性听力丧失或突发性听力丧失。最严重的功能后遗症之一是双侧感音神经性听力丧失,是由肿瘤自行进展和/或与(外科或放射外科)治疗相关的损害所致。
诊断标准
1、 NFI的诊断标准:
① 6 个或以上 CALMs:在青春期前直径> 5 mm或在青春期后直径>15 mm;
② 2个或以上任何类型的神经纤维瘤或1个丛状神经纤维瘤(plexiform neurofibroma,pNF);
③ 腋窝或腹股沟区雀斑;
④ 视神经胶质瘤(optic pathway glioma, OPG);
⑤ 2 个或以上Lisch结节(虹膜错构瘤);
⑥ 特征性骨病变,如蝶骨发育不良或长骨皮质增厚伴或不伴假关节;
⑦ 有一级亲属(父母、同胞或子女)根据上述标准被诊断为NF1。
凡是符合以上2个或2个以上条件的可诊断NF1型。
2、NF2的诊断标准:
1987年美国国立卫生研究所诊断标准:
①CT和MRI检查提示双侧第8对脑神经肿瘤;
②一级亲属患有NF2,患者自身为单侧听神经瘤;
③一级亲属患有NF2,患者自身有以下病变中的2种:神经细胞瘤、脑膜瘤、胶质瘤和青少年后囊下晶状体混浊。
治疗
1、CAMLs 的治疗
目前尚无针对NF1患者的CALMs系统治疗研究。CALMs主要采用对症处理,如皮损严重影响美观,可采用激光治疗,如调Q激光,其治疗原理是选择性光热作用,如调Q532 nm激光、调Q红宝石激光(694 nm)、翠绿宝石激光(755 nm)及低能量调Q1064 nm激光。调Q激光通常不引起瘢痕,但疗效反应个体差异较大。此外,强脉冲光、点阵激光、皮秒及超皮秒激光也可用于CALMs的治疗。
NF1患者的CALMs一般不必处理,对严重影响美观的皮损,可尝试激光治疗。
2、cNF的治疗
手术切除是cNF治疗的主要手段,CO2激光消融、激光烧灼、电干燥术等也可应用。
手术切除目前仅针对少量、较大且有症状的cNF。手术适应证包括:
①瘤体体积较大,对周围组织造成明显压迫和/或使其功能障碍;
②侵犯其他系统;
③近期瘤体明显增大,怀疑有恶变可能或组织学检查证明瘤体存在恶变;
④瘤体破裂伴有急性大量出血;
⑤瘤体影响外观或疼痛等,影响患者生存质量。
CO2激光消融、电干燥术、激光光凝术及射频消融术,用于治疗瘤体数量较多、严重影响外观的cNF患者。
3、神经纤维瘤的手术治疗
手术指征:
产生临床症状的神经纤维瘤、具有恶变影像学证据的病灶,以及体积过大的肿块(直径>6 cm)进行手术。
手术时机:
手术干预的时机目前仍有争议。有学者主张尽早手术,从而避免眼眶等部位并发症的发生及恶化,例如,失明或脑膜脑膨出。此外,在病灶体积较小时进行干预,完全切除的机会更大,复发率也会降低。
而另一些学者建议等到青春期后,待病变稳定后再进行干预。此外,也有学者主张干预时间应尽量推迟,因手术越早,需进行重复手术的次数越多。
手术方式:手术方式根据切除瘤体的比例可分为全切除/近全切除:>80%/90%,次全切除:50%~80%/90%,以及部分切除:<50%。如果可行,推荐全切除/近全切除。
要与包括眼科医生、神经外科医生在内的多学科团队一起制订手术方案。
4、靶向治疗
司美替尼是一种可诱导肿瘤缩小的口服选择性丝裂原活化蛋白激酶(MEK) 抑制剂,于 2020 年 4 月获得美国FDA 批准,用于治疗2~18岁,有症状和/或进行性、不可手术的NF1相关pNF,推荐剂量为25 mg/m2,每日2次。
司美替尼在NF1患者中最常见的不良反应是肌酸磷酸激酶升高、痤疮样皮疹等。司美替尼不太常见但严重的毒性包括左心室射血分数降低、心肌病和眼部毒性。
除MEK抑制剂之外,另有几类靶向疗法在临床试验中表现为对pNF有效,如多酪氨酸激酶抑制剂。此外,针对cNF的外用Ras-MEK通路抑制剂也在研发中。
5、神经纤维瘤病慢性疼痛的治疗
目前尚无针对NF疼痛的特异性疗法。
轻度疼痛者可给予非甾体抗炎药;中重度疼痛者可给予阿片类镇痛药,如吗啡、奥施康定等。当肿瘤压迫、侵犯神经引起神经刺激性症状时(短暂性阵发、反复发作的沿神经分布的放射性刺痛、灼痛,常在夜间发作,体位改变、用力排便等都可以诱发疼痛或使其加重),可加用普瑞巴林、加巴喷丁等治疗神经痛的药物,也可通过手术切除肿瘤获得较长期的疼痛控制。
此外,由于NF患者通常合并严重的情绪问题,还应在专业医生指导下给予抗焦虑、抗抑郁药物治疗和生物-心理-社会学治疗。
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言