
病例报道:新生儿枫糖尿症一例
2015-12-30 毛雪,王华. 中国新生儿科杂志
患儿 男,1天,生后半天因“呕吐”入院,诊断为“咽下综合征”,住院治疗2天后出院。生后第5 天因“发热、腹泻,伴反应差、吃奶差”再次入院。患儿系第1 胎第1 产,胎龄38+5周自然分娩,出生体重3 240 g,无宫内窘迫,Apgar评分1、5、10 min均为10分,其母胎膜早破>18 h。 入院查体: 反应欠佳,哭声小,前囟饱满,张力不高,皮肤轻度黄染,双肺呼吸音粗,腹软,肝脾肋下未
患儿 男,1天,生后半天因“呕吐”入院,诊断为“咽下综合征”,住院治疗2天后出院。生后第5 天因“发热、腹泻,伴反应差、吃奶差”再次入院。患儿系第1 胎第1 产,胎龄38+5周自然分娩,出生体重3 240 g,无宫内窘迫,Apgar评分1、5、10 min均为10分,其母胎膜早破>18 h。
入院查体: 反应欠佳,哭声小,前囟饱满,张力不高,皮肤轻度黄染,双肺呼吸音粗,腹软,肝脾肋下未及,四肢肌张力高,原始反射减弱。生后第7 天出现抽搐,表现为角弓反张、四肢强直,先后给予苯巴比妥、水合氯醛、咪唑安定止惊,仍有反复抽搐。
实验室检查: 血气: pH 7.26,PCO224.75 mmHg, PO287.75mmHg, K+3.8 mmol/L,BE-14.2 mmol/L,HCO3-11.2 mmol/L。肝功能:ALT 23 U/L,AST 30 U/L,TB 169.1 μmol/L,血氨:59.3 μmol/L,血常规: WBC 9.3×109/L,N 55.6%,L 46%,Hb 167 g/L,PLT 203×109/L,CRP 4 mg/L。凝血功能: PT 13.5 s,APTT 47.2 s,Fg 97 mg/dl。两次脑脊液常规、生化、涂片、培养均未见明显异常。TORCH IgM均阴性,粪便腺病毒阴性。
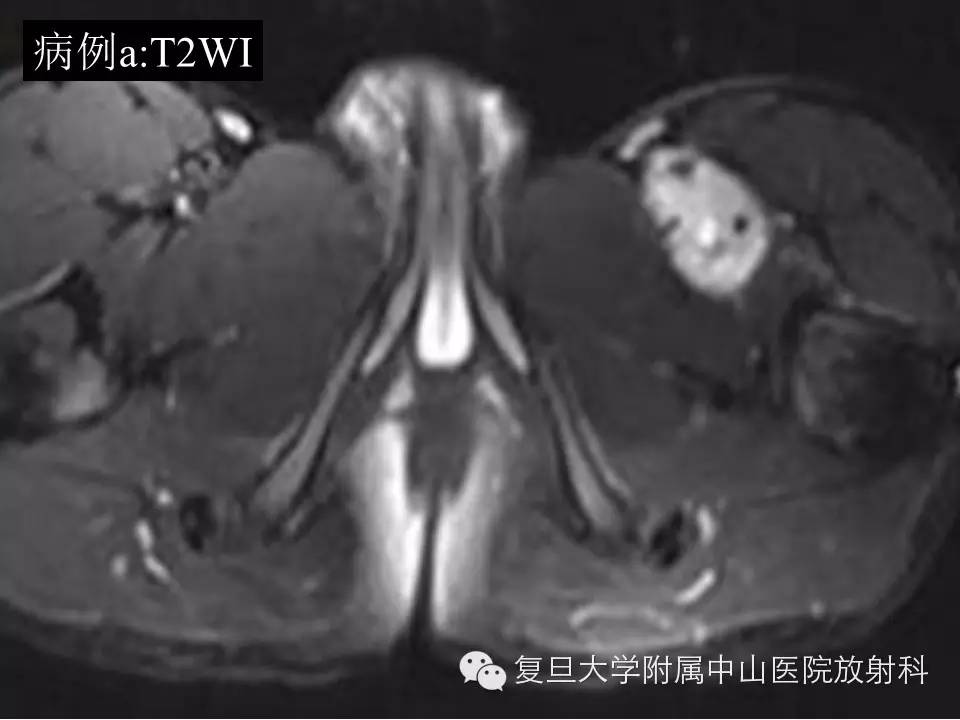
头颅CT: 脑白质密度降低;头颅MRI: 双侧苍白球、丘脑背侧、中脑导水管周围灰质及小脑齿状核信号异常,双侧内囊后肢及脑白质髓鞘未见形成,小脑萎缩。视频脑电图: 异常,额、中央、枕、中颞区多次尖波发放。我院血代谢筛查: 亮氨酸3 068.4 μmol/L( 正常53~230 μmol/L) ,缬氨酸472.09 μmol/L (正常46~230 μmol/L) ,提示枫糖尿症。上海儿童医院尿代谢筛查: 2-羟基异戊酸、2-酮-异戊酸、2-羟基-异葵酸、2-酮-3-羟基戊酸、2-酮-异己酸升高,支持枫糖尿症。
上海市儿科医学研究所内分泌、遗传代谢病研究室基因检测结果: 患儿为可疑复合杂合突变,父母为携带者,父亲突变位点c. 410C>T( p.A137V),已有文献报道为致病突变;母亲突变位点c.2T > C( p.M1T) ,未见报道。
明确诊断后给予免支链氨基酸奶粉+ 母乳3∶1混合喂养,维生素B1 100 mg/( kg·d) 口服,改为左乙拉西坦止惊,惊厥次数及发作形式较前明显减轻,生后25 天亮氨酸降至1312.59 μmol/L,住院40天,出院时患儿吸吮能力较差,奶量80 ml/次,q3h,仍偶有四肢小抽动,出院时体重3.95 kg。院外长期在门诊随访,现生后3个月,体重7 kg,无抽搐发作,吃奶好,特殊奶粉+ 母乳(3∶1),120 ml/次,q2h喂养,查体肌张力高。目前口服复方东维力1/3支,每日3次,已停用左乙拉西坦及维生素B1。复查血亮氨酸700 μmol/L,肝功能恢复正常。视频脑电图:界限性婴儿期脑电图,额、中央区尖波数次发放。头颅MRI与旧片相比苍白球和小脑异常信号消失或明显减少,丘脑背侧、中脑导水管周围灰质及小脑异常信号消失。
讨论
枫糖尿症是一种罕见的支链氨基酸代谢异常疾病,为常染色体隐性遗传。该病是由于支链氨基酸α-酮酸脱氢酶复合体( BCKDH) 基因缺陷,使组织中支链氨基酸和支链α-酮酸异常增高,引起神经系统损害,并排出带有枫糖浆香甜味的尿液。患儿出生时多正常,逐渐出现呕吐、喂养困难、反应低下、意识障碍、肌张力障碍、惊厥、中枢性呼吸困难。血浆中别异亮氨酸高于5μmol/L 被认为是诊断各型枫糖尿症最特异而且敏感的指标。急性期常见弥漫性脑水肿,如果治疗及时水肿改变可逆; 慢性期见对称性基底节、丘脑、齿状核、大脑脚部位损害;约半数患儿可见低血糖及酮尿,血及脑脊液中乳酸水平常可升高,高氨血症并不常见。
治疗分为急性期和慢性期。急性期治疗主要为控制血亮氨酸浓度,如血液净化,对于维生素B1有效型患儿需给予大剂量维生素B1口服。慢性期的特殊饮食治疗应贯穿终生。肝移植术可有效治疗枫糖尿症,但神经系统症状改善不明显。理想控制目标是血亮氨酸浓度为150~300 μmol/L,血异亮氨酸水平与亮氨酸水平相近,血缬氨酸水平至少是亮氨酸的2倍。
该病总体预后不佳。若得到早期诊断,予以正确治疗和预防,可大大降低致残率和病死率。本例患儿经过治疗后神经系统症状、体征及辅助检查有明显好转。本病一旦确诊,先证者及双亲应进行基因监测以明确致病等位基因位点。如果患儿的父母需要再生育,可以在妊娠10~12周后行绒毛膜绒毛取样获得胎儿细胞,或妊娠15~18周后通过羊水穿刺进而得到胎儿的DNA样本,进行产前基因诊断。对于未知致病等位基因位点的病例则可以通过检测体外培养的羊水细胞或者绒毛膜绒毛细胞的BCKDH 活性来进行产前诊断。
原始出处:
毛雪,王华.中国新生儿科杂志,2015年4期:299-300
小知识:枫糖尿症
枫糖尿症(maple syrup urine disease)是常隐遗传的代谢病,首先由Menkes等发现。枫糖尿症是一种罕见的支链氨基酸代谢异常疾病,该病是由于支链氨基酸α-酮酸脱氢酶复合体( BCKDH) 基因缺陷,使组织中支链氨基酸和支链α-酮酸异常增高,引起神经系统损害,并排出带有枫糖浆香甜味的尿液。迄今至少报道有10种类型。各类型普遍都存在支链氨基酸降解酶的缺乏,并早期出现智能发育迟滞和其他神经症状。由于尿中排出的代谢产物具有类似枫糖浆的特异气味,因而命名。
发病率约1/100,000~1/300,000活产儿,我国自1987年起有报道
大致可分四种主要类型,还有罕见的第五型
1.经典型枫糖尿症
患儿在出生时状况良好,一般从生后几天或1-2个月内发现喂养困难,啼哭声弱,不能吸乳和反应迟滞,以后即逐渐消瘦,智能低下,同时呼吸变浅,间断出现发绀现象。体检时可见全身肌张力减低或增高,Moro反射减弱或消失,强直性惊厥或角弓反张等都较常见。小儿囟门常膨出而紧张,还有时出现眼震、眼肌瘫痪、睑下垂、瞳孔散大及对光无反应等。病情进展迅速,尿中逐渐出现特异气味。本型在文献报告的病例中约占70%,一般症状较严重,预后较差,多死于酮中毒。
2.间歇性枫糖尿症
早期发育正常,反应也不迟钝,大约从生后10个月到2岁间歇性出现厌食、呕吐、表情淡漠、步态不稳、共济失调、嗜睡和行为改变等。尿中有特异的气味。病程长短不一,可以有多次起伏,也有的患儿发生急性酮中毒而死亡。本型约占20%。
3.轻型枫糖尿症
表现为精神发育迟滞,但无其他典型神经症状和体征,也没有间歇发作的特点。
4.对VB1反应型枫糖尿症
仅有轻度智能发育迟滞,也无典型的或间歇神经损害症状。仅有血中支链酮酸的含量比正常儿稍高。本型患儿对维生素B1的疗效好,血中的生化异常可有显著的好转。
5. (E3)缺乏型枫糖尿症
极为罕见,临床表现类似中间型,但由于E3亚单位的缺陷,患儿除支链α-酮酸脱氢酶活力低下外,其丙酮酸脱氢酶和α-酮戊二酸脱氢酶功能亦受损,故伴有严重乳酸酸中毒。患儿在出生数月内通常不出现症状,随着病程进展,逐渐出现进行性的神经系统症状,如肌张力减低、运动障碍、发育迟滞等。尿液中大量排出乳酸、丙酮酸、α-酮戊二酸、α-羟基异戊酸和α-羟基酮戊二酸等;由于丙酮酸的大量累积,血中丙氨酸浓度亦增高。本型患儿限制蛋白和脂肪摄人,应用大剂量VB1等治疗均无效。
病理
病儿神经系统正常发育过程受阻,大脑皮质除海马以外各部位层次不清,细胞结构也不成熟。神经母细胞的外向移动受阻,以致皮质以外有异位灰质存在(常见于脑室的室管膜周围),说明在妊娠后期1/3中枢神经系统的发育有明显受阻。其次,几乎所有中枢神经的髓鞘化过程都受损,在大脑半球白质、锥体束、小脑齿状核和胼胝体等部位尤其明显,仅有内囊、视束、嗅束和脊髓后索不受影响。此外,灰白质中有广泛的囊性改变,显示空泡形成,周围有多数星形胶质增生,形成海绵状态。用组化方法染色发现空泡内含PAS阳性物质,但未发现髓鞘裂解的产物。
实验室检查
用色谱分析法可测出经典型患儿血中几乎所有的支链氨基酸含量都明显增高(如亮氨酸平均增高30倍,异亮氨酸平均增高16.5倍,缬氨酸平均增高8 倍),有时还在血中发现异亮氨酸的立体异构物别异亮氨酸(alloisoleucine),其他的氨基酸无大变化,同时还可测出血浆和尿中支链酮酸 (NICA、KIVA、KMVA等)也大量增多,尿中a-羟基丁酸和a-羟基异戊酸等也常增高。在细胞培养中,可发现KICA和KMVA脱羧基酶活性显著降低,KIVA脱羧基酶活性也可下降。此外,患儿还经常出现低血糖(空腹血糖有时低于600mg/L),葡萄糖耐量曲线呈低平型,可能是继发于糖代谢障碍,血中胰岛素过高或者糖原异生作用受损的关系。并有血中pH、pC02、HCOs含量下降,尿酸、尿中吲哚乙酸类排出增多等继发的代谢紊乱。
治疗原则
本病的治疗原则是膳食控制。在患儿的食品中严格限制支链氨基酸类,蛋白总量也需限制在每日2g/kg体重以下,并需经常检查血中的氨基酸浓度来参照。患儿在感染、外伤或有肠胃疾病时,由于代谢性失代偿作用而致症状加重。因此,此时必须采用碱性溶液矫正酸中毒,减少蛋白人量和保持负氮平衡,必要时需反复输血或腹腔透析以挽救生命。此外,本病应常规应用维生素B1每日10~20mg,至少维持2~3年。对于产前诊断为阳性的胎儿,应尽可能地劝告其终止妊娠。
【附】其他有关的支链氨基酸代谢病
1. 高缬氨酸血症
首先由和田(Wada)等(1965)报告的罕见代谢病。其代谢缺陷可能是缬氨酸转氨基酶的活性缺乏。临床表现与枫糖尿症相似,也有难喂养、呕吐,不能吸乳等特点,但并不嗜睡,且常出现眼震,和枫糖尿症不同。患儿血和尿中缬氨酸增高,而其他氨基酸含量正常。通过限制支链氨基酸膳食可改善临床症状。
2.异戊酸血症
首先由田中(Tanaka)等(1966)报告的脂肪酸代谢障碍。病因可能是亮氨酸降解过程中异戊酰辅酶A脱氢酶的活性缺乏,以致其底物异戊酸在体内蓄积。临床特点是患儿全身有特异的"干奶酪"或"汗脚"的气味,并表现有发作性酸中毒和昏迷,同时出现不同程度运动功能和智能发育迟滞,有些患儿从新生儿期就出现呕吐和惊厥,并能迅速出现酸中毒而危及生命。实验室检查见血中异戊酸含量明显增高(可达正常婴儿的1500倍)。用C标记可见病儿末梢血中白细胞的1-14C-异戊酸的氧化明显抑制。用亮氨酸内服作负荷试验,也可见血中亮氨酸含量相应上升。治疗方法同上。
3.B-羟基异戊酸(B_hydroxyisovaleric)和甲基丁烯酰甘氨酸尿症
这是另一种亮氨酸分解的代谢病,首先由Eldjarn等(1970)和Stokke等(1972)描述。代谢缺陷可能是甲基丙烯酰辅酶A羧化酶活性缺乏。临床表现是出生2周左右出现上下肢肌张力低,运动发育迟滞。以后逐渐出现肌萎缩、腱反射缺失和舌肌纤颤等,与婴儿型脊性肌萎缩(Werdnig- Hoffmann病)相似。患儿的尿常有特异的"猫尿"气味,有时有助于诊断。在实验室检查中,尿液可测出大量的B_羟基异戊酸(HIV)和B_甲基丁烯酰甘氨酸(MCG)。从患儿白细胞或成纤维细胞培养中可测出酶活性。治疗方面也以限制亮氨酸膳食为主(每日不超过150mg/kg体重),由于生物素是羧化酶辅酶,每日内服生物素250mg,可使患儿尿中排出的有机酸减少,但对临床症状有无疗效尚未肯定。
4.甲基-p羟基丁酸尿症
同属亮氨酸分解障碍,首先由Daum等(1971)报告。其代谢缺陷可能是硫解酶的活性缺乏,不能使亮氨酸的降解物a-甲基乙酰辅酶A转化为丙酰辅酶 A和乙酰辅酶A。患儿的临床表现为反复发作的代谢性酸中毒和智能发育迟滞,严重者伴意识障碍。尿中排出大量a-甲基-B羟基丁酸(MHBA),但无特异气味。治疗原则以限制蛋白摄入量(每日<2g/kg体重=为主。
作者:毛雪,王华.
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





以前见过一些,但是很多年轻医生都没有听过这个疾病,更难以说就可以确诊,最后患儿往往有好不好。
166
不错哦
127
不错哦
115
不错哦
114
赞一个
150