你知道克雅病吗?
2017-10-10 张微微 戚晓昆 神经科少见病例
克雅病(Creutzfeldt‐Jakob disease,CJD)具有特殊条件下的传染性,极少的有遗传性,多数为散发。目前尚无治疗手段,确诊依赖病理学检查。在脑活检受各种因素影响不能广泛开展的情况下,掌握CJD临床及辅助检查特点,对于减少误诊具重要的意义。现报道1例符合“全国CJD监测方案散发型CJD临床诊断标准”的病例。
克雅病(Creutzfeldt‐Jakob disease,CJD)具有特殊条件下的传染性,极少的有遗传性,多数为散发。目前尚无治疗手段,确诊依赖病理学检查。在脑活检受各种因素影响不能广泛开展的情况下,掌握CJD临床及辅助检查特点,对于减少误诊具重要的意义。现报道1例符合“全国CJD监测方案散发型CJD临床诊断标准”的病例。
临床资料
患者男性,61岁。内蒙籍。主因发作性眩晕5周、步态不稳、肢体抖动、视物变形4周于2007年11月1日入院。
2007年9月下旬,患者无明显诱因出现阵发性眩晕,多在坐、卧位改直立位时出现,持续1小时左右可缓解,无发热、头痛或呕吐。当地医院按“脑梗死”治疗无效。10月初出现步态不稳(醉酒步态),同时言语不流利、视物变形、双手动作不准,肢体间断抖动(入睡后明显,以左侧肢体为重)。上述症状进行性加重,10月下旬出现时间地点定向障碍,间断幻视、幻听、欣快、食欲进行性减退。既往无高血压、手术或激素应用史。
入院查体:血压140/90mmHg。皮肤、浅表淋巴结、心、肺、腹部未见异常。神经系统:神清,吐字有爆发倾向(有爆破样语言)。交流初欣快(开始交流时有欣快感),数分钟后沉默寡言。远、近记忆力明显减退,计算力差。时间、地点定向障碍。脑神经及肢体未见瘫痪。感觉无缺失(无感觉缺失)。四肢肌容积正常、肌张力增高,共济动作不准确(共济动作欠稳准),左上肢可见肌阵挛及静止性震颤。四肢腱反射(++),病理反射未引出。颈软,脑膜刺激征(-)。
入院后检查:血常规、尿常规、大便常规、肝功能、肾功能、血糖、血脂、血离子、心酶正常;HIV、梅毒反应、HBsAg、HCV(-),ASO、RF、ESR、免疫球蛋白正常。心电图、胸片、心脏超声、消化及泌尿系统超声未见异常,头颅CT未见异常,脑血管MRA未见异常。
脑脊液(2007‐11‐5):无色透明,初压120mmH2O,白细胞1、红细胞1(×106/L),糖、蛋白及氯化物正常、未见肿瘤细胞。血与脑脊液结核卡、单孢病毒检测(-)。血神经元特异性烯醇化酶(NSE)28.58μg(正常值0~15.2μg)。
脑电图:慢波背景,右额颞叶间隔0.8秒左右一次45~60毫秒、50~70mV尖波(图4.4‐1)。头颅增强MR:DWI序列示右侧尾状核,额颞叶皮质及皮质下信号较对侧增高,双侧底节、脑室旁、半卵圆中心可见多片T1稍低、T2FLAIR稍高异常信号(图4.4‐2)。
入院后无特殊治疗。患者病情进行性加重,住院第7天意识蒙,无动性缄默,不能进食水,尿潴留。
2007年11月7日复查脑脊液,无色透明,压力、常规、生化结果正常。脑脊液14‐3‐3蛋白Western Blot检测(+),血PRNP全序列无突变,第129位氨基酸多态性为M/M型。
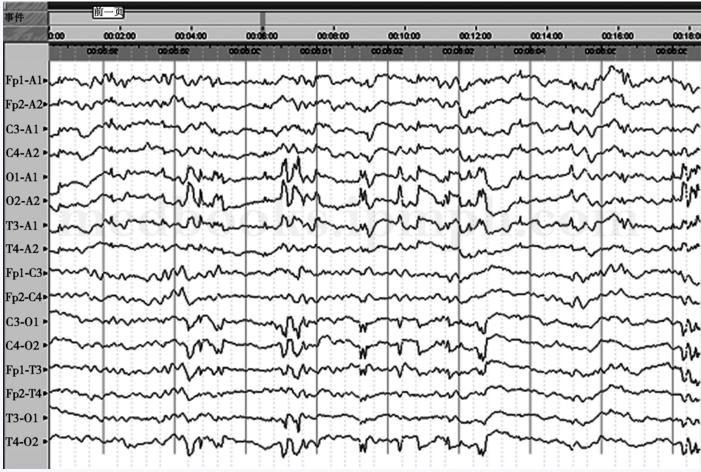
慢波背景,右额颞叶间隔0.8秒左右1次45~60毫秒、50~70mV尖波
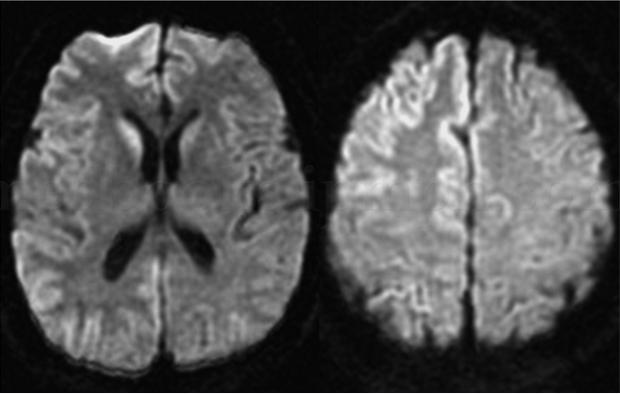
DWI序列示右侧尾状核,额颞叶皮质及皮质下信号较对侧增高,双侧基底节、脑室旁、半卵圆中心可见多片T1稍低、T2FLAIR稍高异常信号。
讨论
CJD是人类最常见的可传播性海绵状脑病(TSE)、称为皮质‐纹状体‐脊髓变性。由德国神经病学家Creutzfeldt和Jakob分别于1920年和1921年首先报道,1922年首次使用了“克雅病”。年发病率约为百万分之一。临床分为散发型(85%,平均病程15个月)、遗传型(5%~15%,平均病程23个月)、医源型和变异型,是中枢神经系统致死性退行性疾病。变异型CJD可以通过血液传播[1]。
CJD的发生与编码朊粒的基因(PRNP)突变、PRNP翻译错误或感染外源性羊瘙痒病朊粒(PrPSC)有关[2]。PrPSC在脑内大量沉积所引起广泛神经细胞丢失,脑组织呈现海绵样变、PrP淀粉样斑块形成是CJD的确诊标准。
CJD临床特点是快速进展的锥体外系症状及认知功能障碍,平均病程11个月。本例以小脑综合征为首发症状,随后出现快速进展的认知功能障碍、肌阵挛,病程60余天发展为无动性缄默,与CJD的临床特点相符合。
脑电图的特征是间隔0.5~2.5秒重复出现高幅尖波、慢波背景上出现广泛双相或三相周期性尖慢复合波,对CJD诊断的敏感性为67%[3]。本例脑电图慢波背景,右额颞叶出现周期性尖波,较为符合。
大脑皮质和(或)纹状体的MRI异常高信号是CJD的影像学特征,79%的散发性CJD患者双侧尾状核、壳核呈对称性长T2信号,92%的变异性CJD可见双侧丘脑对称性的长T2信号。DWI更为敏感,异常高信号改变在疾病早期常不对称,随病程进展趋于对称。DWI异常信号最早出现在病后1个月,常先于脑电图及脑脊液的异常变化,对CJD诊断的敏感度及特异度可达到100%[4]。本例MRI改变右侧尾状核、额颞叶皮质及皮质下异常DWI信号,与徐全刚等[5]介绍的CJD特征性影像学改变相符,具有诊断价值。
1967年Moore和Perez对脑组织电泳时发现一系列酸性蛋白质,根据其在层析及电泳凝胶中的迁移位置命名为14‐3‐3蛋白,主要分布在神经细胞内,约占全脑可溶性蛋白的1%,参与细胞分裂周期和细胞凋亡的信号转导。正常情况下脑脊液中不含14‐3‐3蛋白,而在神经退行性病变,尤其CJD时脑脊液含量显著升高[6]。CSF采样时患者临床诊断疑似程度与14‐3‐3阳性预测值正相关,西班牙1068例临床可疑sCJD患者,按WHO诊断标准(不包括14‐3‐3检测)分为可能(probable)166例、疑似(possible)129例和非sCJD(non)773例,14‐3‐3蛋白测定阳性预测值分别为98.4%、97.5%和31%,阴性预测值分别为22.2%、73.4%和100%[7]。本例脑脊液14‐3‐3蛋白Western Blot测定阳性,支持诊断。
PRNP有50多个位点的突变可导致不同类型的CJD,大多数散发型CJD与129位点的甲硫氨酸(Met)纯合子有关。利用聚合酶链反应技术测定DNA产物序列,在编码129区观察到纯合子(M/M)是诊断CJD的有力证据。本例PRNP全序列测定与标准序列比对序列无突变,129位氨基酸多态性为M/M型,提示易于感染。
点评
1﹒散发型CJD是人类最常见的致死性可传播性海绵状脑病。
2﹒确认“金标准”基于脑组织活检,但实施困难。
3﹒临床诊断重要依据是快速进展的智能减退、肌阵挛、周期性同步异常EEG、影像可见皮质基底神经节区不对称性异常DWI高信号、CSF阳性或阴性14‐3‐3蛋白。
4﹒患者的神经组织、CSF和血液可能具有传染性。
作者:张微微 戚晓昆
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
小提示:本篇资讯需要登录阅读,点击跳转登录




#克雅病#
51
非常好的资料.学习了
62
学习了.谢谢
63
克雅病(Creutzfeldt‐Jakobdisease.CJD)具有特殊条件下的传染性.极少的有遗传性.多数为散发.
51
学习扩展一下
55