
Nat Genet:eQTL-GWAS分析新算法cTWAS发现调控复杂性状的风险基因
2024-03-08 测序中国 测序中国
研究团队提出了一种用于联合eQTL-GWAS分析的新方法cTWAS(causal-TWAS)。
全基因组关联研究(GWAS)已经确定了一系列与人类性状相关的基因位点。但大多数人类疾病并不是由单一的遗传变异引起的,而是多种基因、环境因素和其他变量复杂相互作用的结果,因此,确定位点如何影响复杂人类性状的机制仍然具有挑战性。
表达数量性状位点(eQTL)是一类能够影响基因表达量的遗传位点,目前已开发出许多方法利用eQTL数据来挖掘GWAS的候选基因,例如共定位分析、全转录组关联研究(TWAS)和孟德尔随机化法。但这些方法都存在一个关键问题:当利用基因的eQTLs评估基因在性状中的作用时,附近的变异和其他基因表达的遗传组件可能与这些eQTLs相关,并对性状产生直接影响。现有方法未能考虑到这些"遗传干扰因素",导致了假阳性。
美国达特茅斯学院赵思明联合芝加哥大学贺信、Matthew Stephens研究团队在Nature Genetics上发表了题为“Adjusting for genetic confounders in transcriptome-wide association studies improves discovery of risk genes of complex traits”的文章。为解决现有方法的局限性,研究团队提出了一种用于联合eQTL-GWAS分析的新方法cTWAS(causal-TWAS)。cTWAS借鉴统计学精细定位的特点,调整了所有的遗传干扰因素。该模型不只关注一个基因,而是考虑了多个基因和变异,并使用贝叶斯多元回归模型,剔除混杂的基因和变异。在真实数据的实际模拟和应用中,cTWAS大大减少了TWAS、共定位和基于MR方法的错误发现数量,为从GWAS中可靠地发现因果基因奠定了基础。

文章发表在Nature Genetics
TWAS面临的关键挑战是,已鉴定的基因对表型并非总是有因果效应,也会有“水平多效性”(图1a)。联合分析eQTL和GWAS数据的方法也面临类似的挑战。为了控制所有潜在的混杂因素,cTWAS联合模拟了表型对所有估算基因和所有变异的依赖性(图1c),所有这些参数的联合估计将导致因果效应估计。cTWAS中变量之间潜在的高相关性提出了新的挑战。因此,研究人员使用为精细映射而开发的统计机制来拟合cTWAS,并使用经验贝叶斯策略来估计这些先验参数,可以同时注释多个基因和变异。根据估计的参数推断出每个区块中可能的因果基因和变异(图1d)。
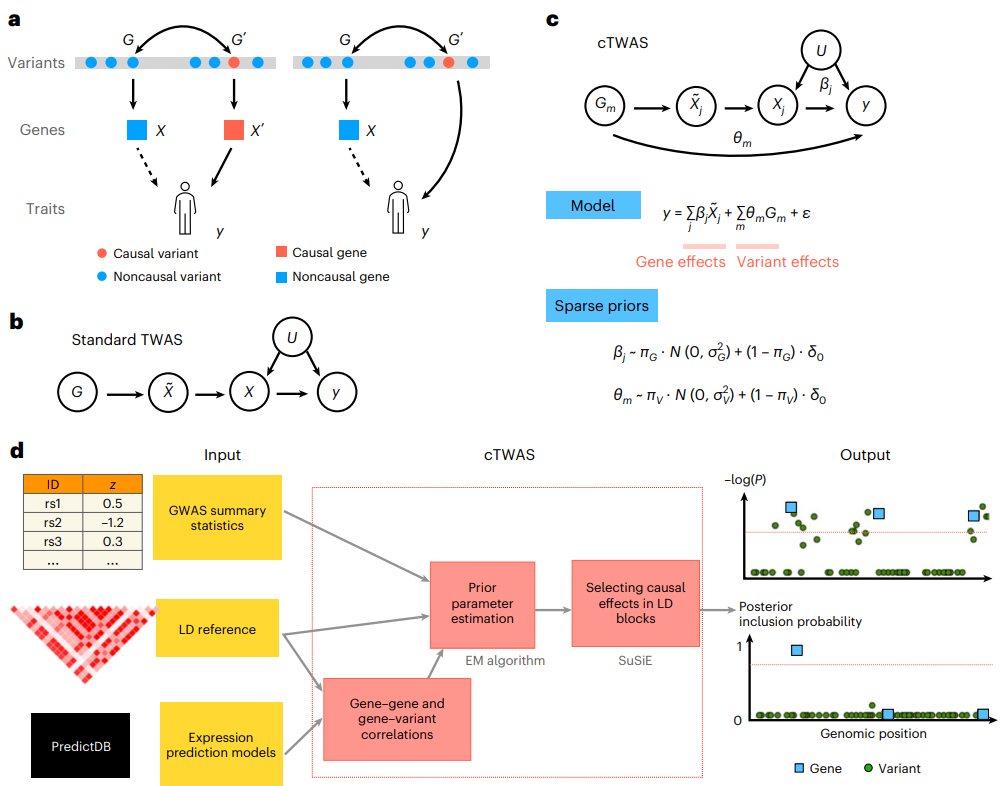
图1.cTWAS方法概述
cTWAS能减少模拟研究中的错误发现
研究团队设计了真实的模拟来评估cTWAS的性能,根据已有研究的真实遗传参数,创建了所有区域的全基因组数据。具体包括使用英国生物样本库约45k份英国白人祖先样本中次要等位基因频率>0.05的变异基因型数据,并通过FUSION利用GTEx的预测模型估算了基因表达,改变了基因和单核苷酸多态性(SNPs)因果关系的先验概率,以及先验效应大小方差。cTWAS估计的参数接近真实值,并准确地估计了由基因效应解释的性状变异的比例(图2a )。研究还发现,通过cTWAS计算基因的PIPs得到了良好校准,预计超过阈值的基因中至少有90%是因果基因。
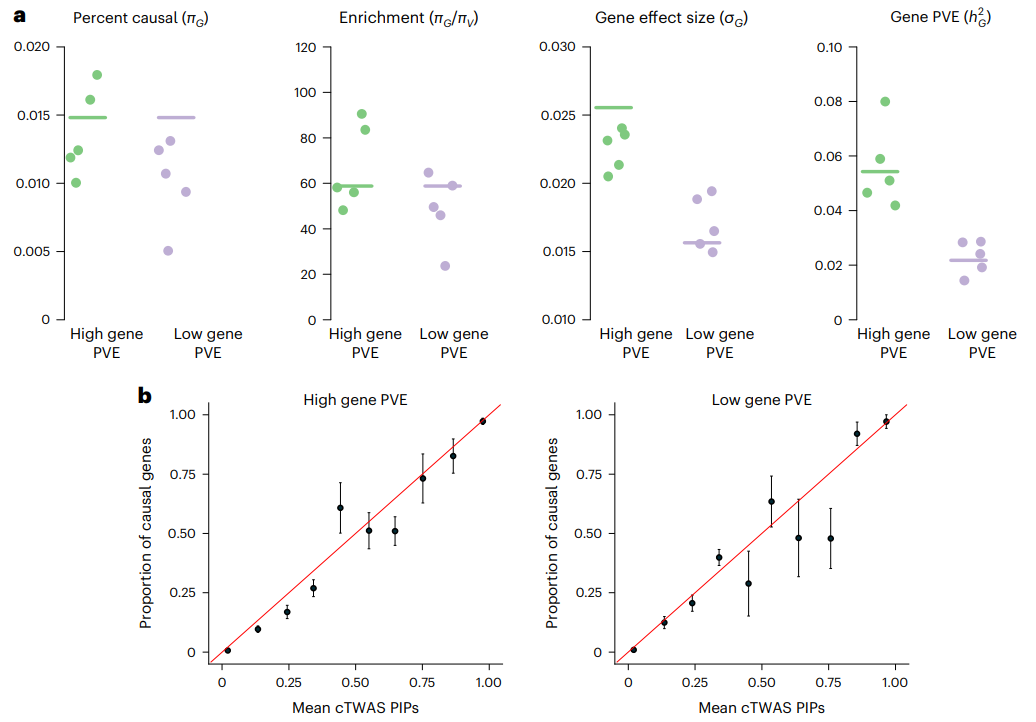
图2.模拟参数评估和PIP校准
研究人员将cTWAS的性能与其他方法进行了系统比较。相比之下,cTWAS成功地去除了许多标准TWAS中具有高度显著相关性的非因果基因(图3a),控制了所有情况下错误发现的比例(图3b),虽然阈值PIP>0.8,但实际的错误发现比例远低于20%。此外,研究人员举例说明了cTWAS是如何消除假阳性的(图3c、d),并且cTWAS对不同的模拟设置具有鲁棒性。
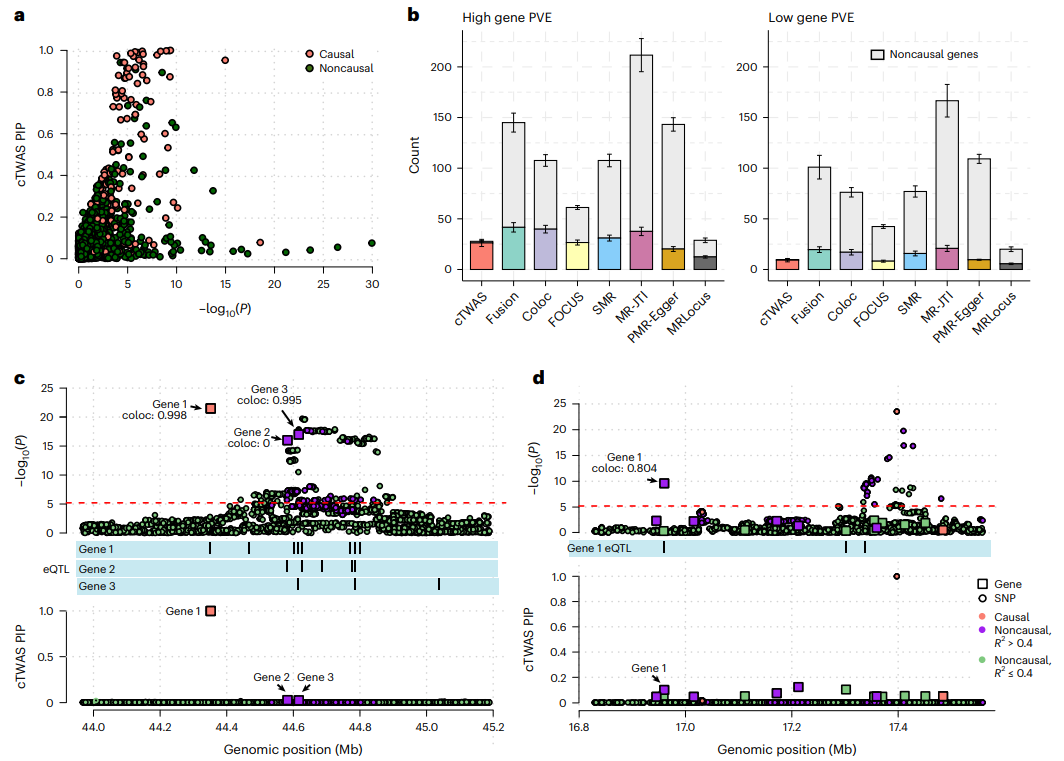
图3. cTWAS与其他模拟方法的对比
cTWAS分析LDL相关致病基因
研究人员将cTWAS应用于英国生物样本库的低密度脂蛋白(LDL)胆固醇的GWAS分析,鉴定出35个PIP>0.8的基因。LDL相关基因分析显示,cTWAS的精度为75%(图4a),大大优于标准TWAS,后者的精度为31%。
研究人员利用两个基因位点说明了cTWAS如何避免假阳性。第一个基因位点包括HPR和其他四个基因。第二个基因位点有3个通过TWAS与LDL相关的基因。结果表明,cTWAS能够避免假阳性基因(图4c)。此外,研究人员系统地评估了标准TWAS假阳性结果的来源,发现TWAS的最大风险不是附近基因之间共享的eQTL,而是基因与附近变异的相关性,这些变异的影响不表现为eQTL。
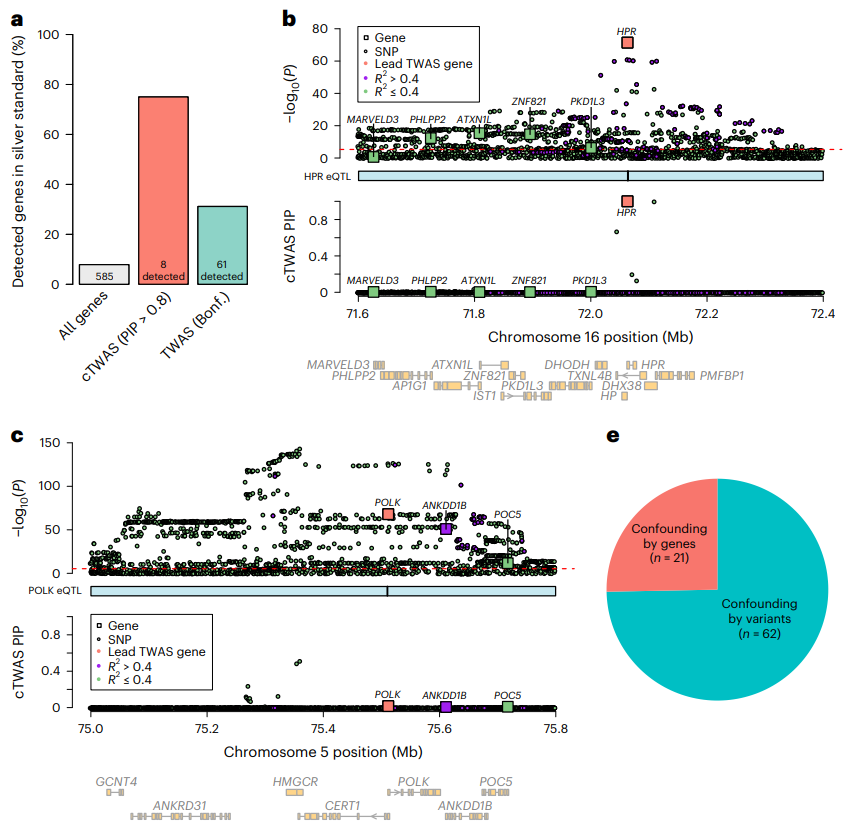
图4.cTWAS准确鉴定了LDL胆固醇的致病基因
cTWAS发现几种常见性状的候选基因
研究团队将cTWAS应用于炎症性肠病(IBD)、收缩压(SBP)和精神分裂症(SCZ)的GWAS汇总统计,使用了GTEx 49个组织的蛋白质编码基因表达预测模型,对每个组织分别进行了cTWAS分析。研究显示,将多个组织的结果结合起来可增强cTWAS的发现能力。此外,cTWAS发现了一些新基因,即IFNGR2、FOSL2、STAT3、FCGR2A、IRF8和ZFP36L2位于已知的IBD相关基因座内,并具有免疫功能。cTWAS还在基因位点中鉴定出关联性低于标准GWAS临界值的新基因,其中一些基因(UBE2W、TYMP、LSP1和CCR5等)具有IBD相关功能(图5f)。
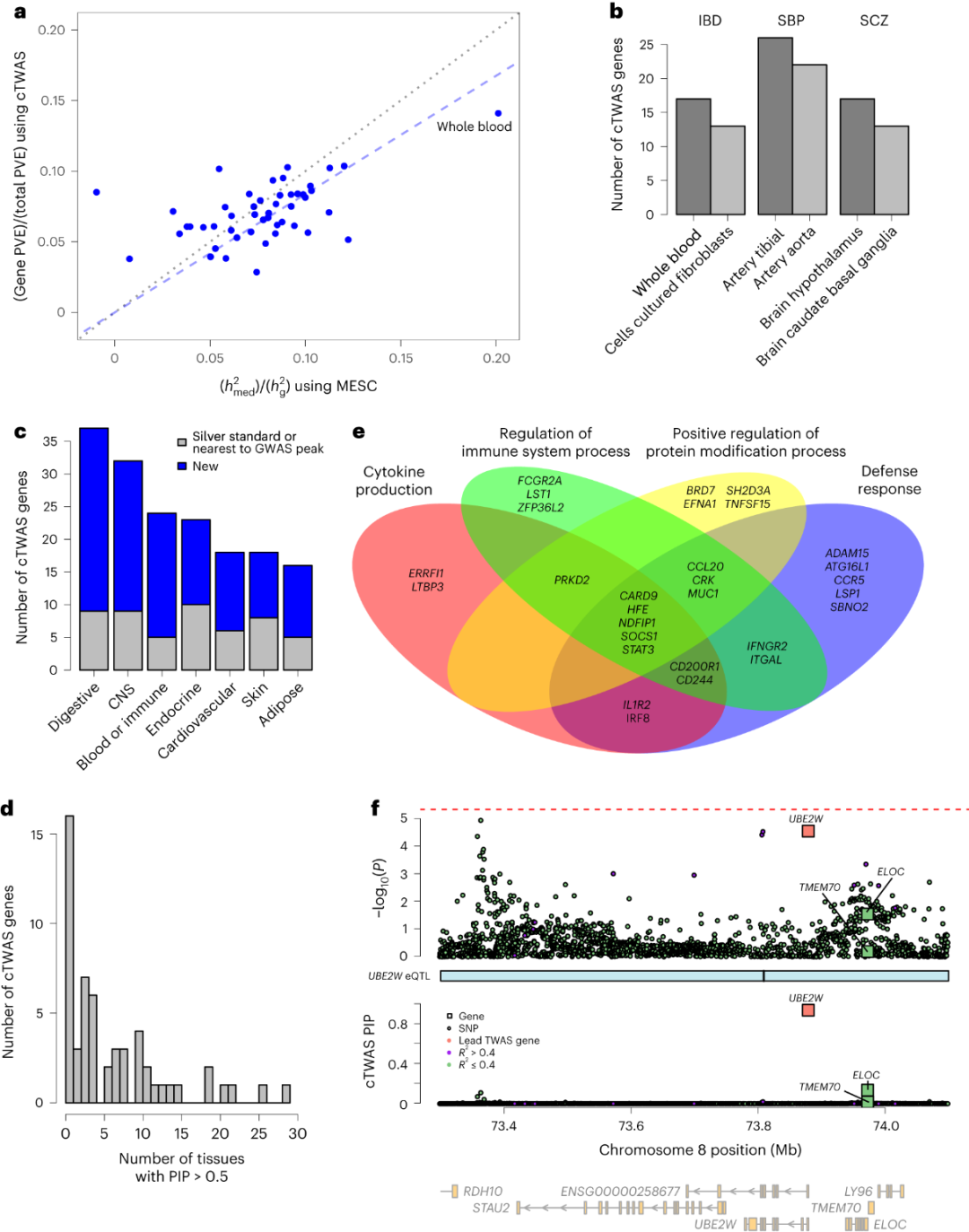
图5.使用GTEx数据库对IBD及其他性状进行cTWAS分析
综上所述, 通过对遗传变异和估算基因表达进行联合建模,cTWAS解释了多效性效应,为检测致病基因创造了一个强大的框架。通过对多个GWAS性状的模拟和应用,cTWAS减少了错误发现,并发现了这些性状的多个候选基因,具有作为基因发现工具的潜力。随着大量分子QTL数据集的可用和生成,cTWAS有望将疾病的遗传关联转化为风险基因、疾病机制和潜在治疗靶点的信息。
论文原文:
Zhao S, Crouse W, Qian S, Luo K, Stephens M, He X. Adjusting for genetic confounders in transcriptome-wide association studies improves discovery of risk genes of complex traits. Nat Genet. 2024. doi:10.1038/s41588-023-01648-9
作者:测序中国
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#全基因组关联研究# #cTWAS#
87