
影像诊断 | 腓骨肌萎缩症(少见、有特点!)
2023-10-26 影像诊断与科研 影像诊断与科研
腓骨肌萎缩症又称遗传性运动感觉神经病,是一组具有高度临床和遗传异质性的周围神经单基因遗传病,发病率约在40/100000。
腓骨肌萎缩症
腓骨肌萎缩症(CMT),又称遗传性运动感觉神经病(hereditary motor and sensory neuropathy,HMSN),是一组具有高度临床和遗传异质性的周围神经单基因遗传病,发病率约在40/100000。主要临床表现为进行性对称性肢体远端肌无力和肌萎缩,感觉障碍和腱反射减退或消失。

CMT多为儿童和青少年起病,典型的临床特点是双下肢最早受累,表现为远端肌肉进行性对称性肌无力,肌萎缩或肥大,腱反射减弱或消失,并伴随骨骼畸形,包括鹤腿样畸形、弓形足以及锤状趾等。患者运动能力出现严重损害,而感觉功能受累程度不一。大多数CMT患者都有一定程度的身体残疾,但很少会影响到涉及呼吸等重要功能的肌肉,因此不会危及生命,大多数CMT患者的寿命与正常人无异。
典型症状
1.四肢远端肌无力:以下肢远端为主,并逐渐向近端发展的肢体肌肉无力。发病开始双脚没有力气、活动不灵、麻木、腓骨肌开始萎缩,后慢慢扩展至骨间肌、小腿屈肌,最后累及大腿下三分之一肌肉,但其上部完全正常,形成“鹤腿”或倒置的酒瓶样畸形。
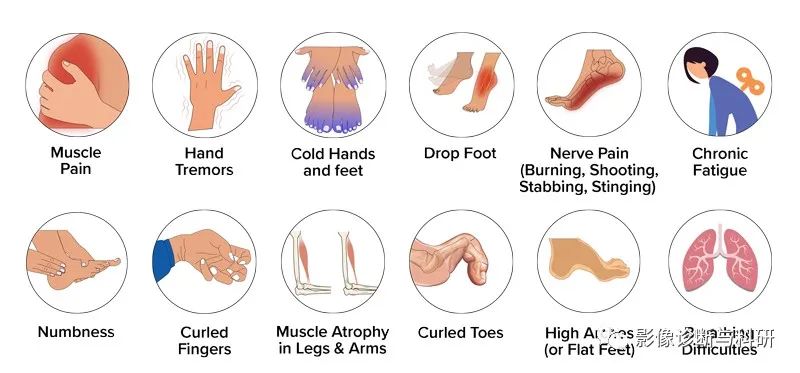
2.肌萎缩:进行性对称性肢体远端肌萎缩,由下肢开始逐渐发展到上肢。萎缩肌肉能有肌束震颤。跟腱反射早期减弱或消失。因为足背屈无力多是马蹄内翻畸形。后面手部出现骨间肌,大、小鱼际肌萎缩,形成猿手畸形,但萎缩一般不超过肘关节以上。

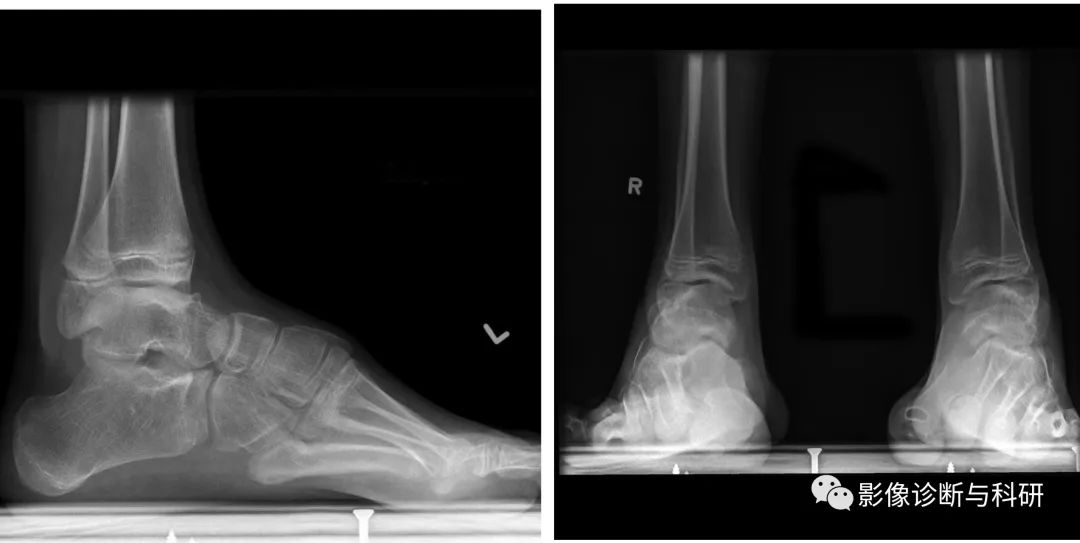
2.肢体感觉障碍:患者有深部感觉减退,出现运动能量差,跑步困难,易扭脚。肢体远端呈套式感觉减退,多有肿胀、紫绀、溃疡等神经营养障碍。
4.其他症状部分患者偶见视神经萎缩、瞳孔改变、眼球震颤及三叉神经痛。
根据临床表现和电生理特征,分为五型:
-
脱髓鞘型(CMT 1):在10岁以内发病慢性进展性病程;周围神经对称性肢体远端肌无力和肌萎缩自足和下肢开始;常伴脊柱侧弯垂足呈跨阈步态;部分病人仅有弓形足或神经传导速度减慢甚至无临床症状。
-
轴索变性型(CMT 2): 发病晚成年开始出现肌萎缩症状及出现部位与CMT1型相似程度较轻;运动NCV正常或接近正常CSF蛋白正常或轻度增高;神经活检主要为轴突变性。
-
CMT 3 :婴儿期起病的严重脱髓鞘性CMT ,即Dejerine-Sottas病(DSD);
-
CMT 4 :大部分隐性遗传性CMT ;
-
CMTX :X连锁遗传 。
-
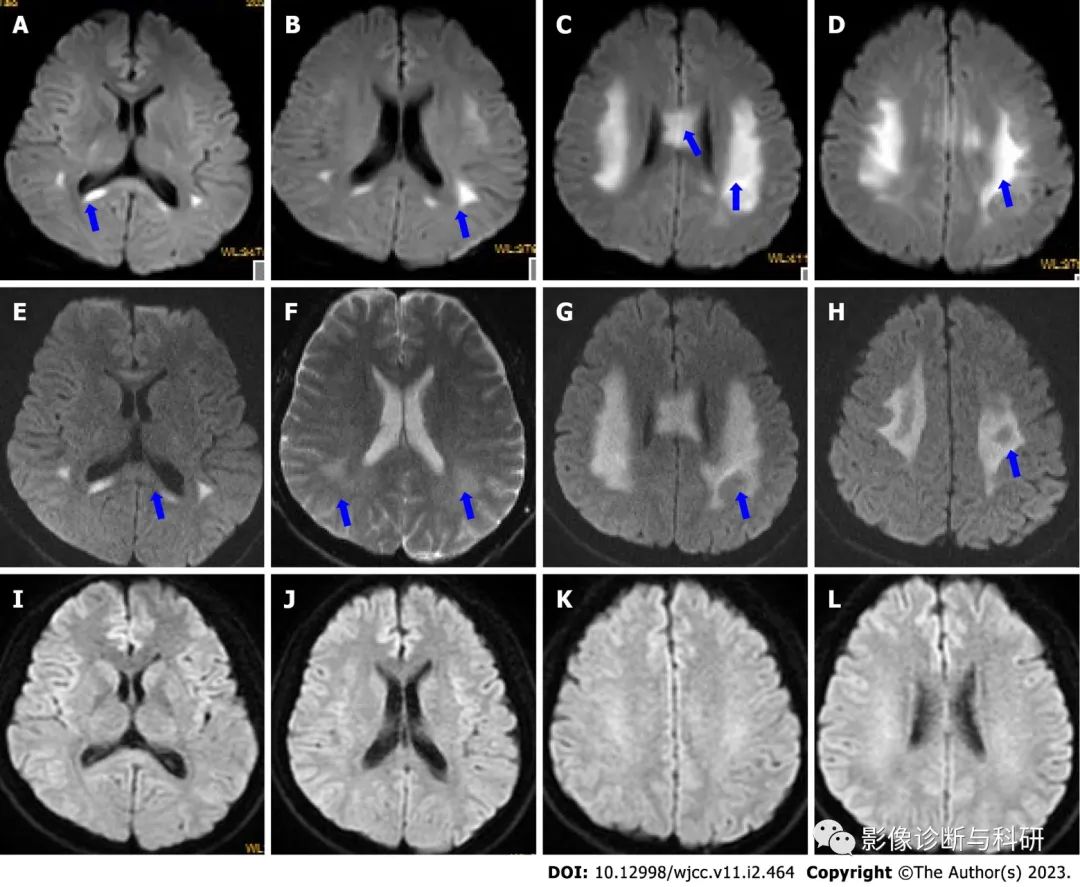
MRI表现为可逆性的双侧胼胝体、放射冠区、内囊后肢的多发对称性长T1长T2信号,FLAIR高信号,弥散受限,无强化。外周神经根肥大。肌电图检查对疾病诊断及鉴别诊断有重要意义,确诊有赖于基因检测。
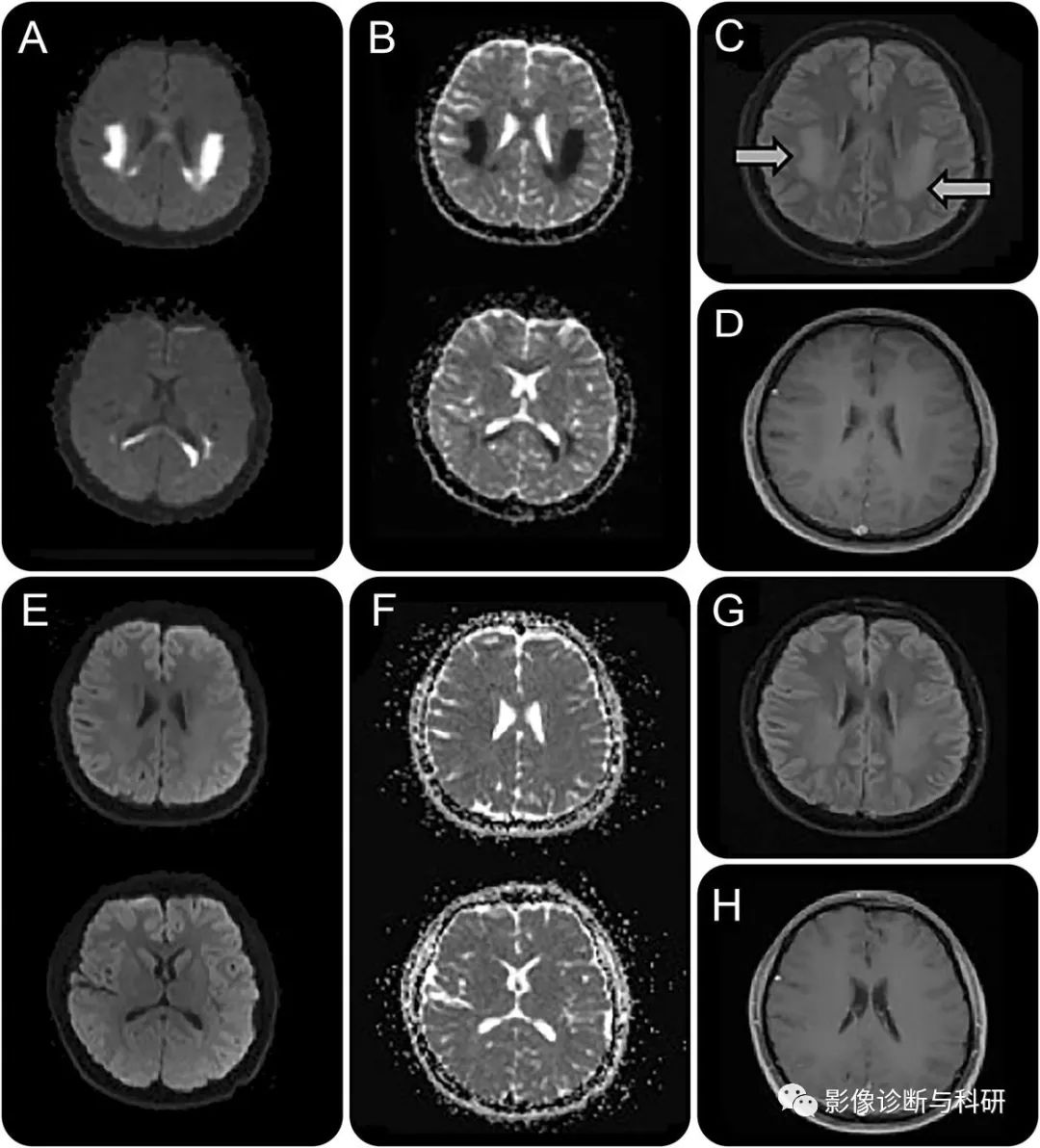
脑部 MRI (A-D) 显示后皮质白质和胼胝体压部的弥散张量序列 (A) 呈高信号,而表观弥散系数序列 (B) 呈相应的低信号。流体衰减反转恢复 (FLAIR) 序列显示出相应的 T2 高信号(方框 C 中的箭头),而钆后 T1 序列显示没有相关的对比度增强 (D)。6 个月随访 (E-H) 时获得的脑 MRI 显示白质脑病几乎完全消退(扩散张量 [E]、表观扩散系数 [F]、FLAIR [G] 和 T1 后钆 [H] 序列所示)。仅持续存在非常轻微的 FLAIR 信号异常 (G)。
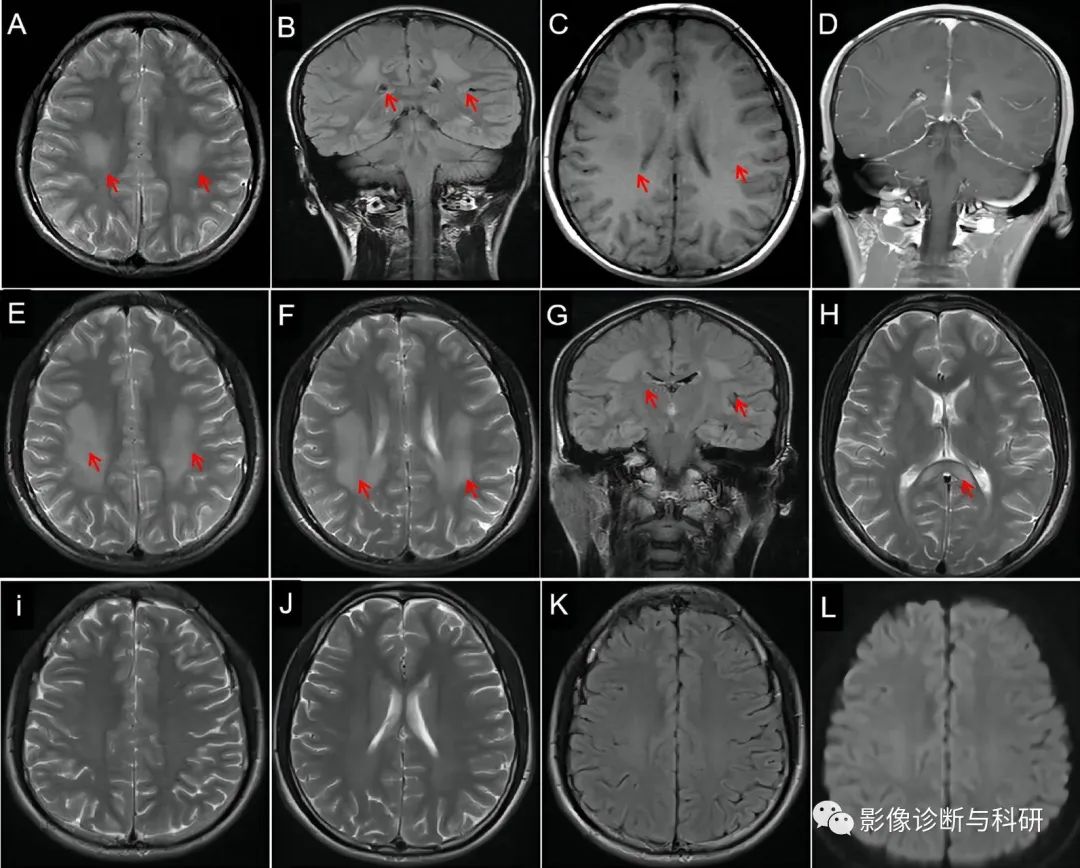
一名8岁男孩于2015年7月因急性发作的言语不清、四肢无力1天入院。脑部磁共振成像 (MRI) 上病变的演变。轴向T2加权成像(WI,A)和冠状T2-WI(B,箭头)上半卵圆中心和放射冠的高信号病变(箭头),T1-WI上的低信号病变(C,箭头),以及造影剂上的等信号增强磁共振成像(D)。MRI在 T2-WI 上显示双侧半卵圆中心( E,箭头)、放射冠(F,G,箭头)和胼胝体(H ,箭头)比以前有更多的高信号。三个月后,重复MRI显示T2-WI上异常白质信号完全消失(I,J)、流体衰减反转恢复成像(K)和扩散 WI (L)。

A:初次头部计算机断层扫描(CT)未显示明显异常;B:第 2 天的磁共振成像 (MRI) 显示 T1 加权成像双侧、对称和明确的低信号(蓝色箭头);C 和 D:表观扩散系数映射 (C) 和 T2 加权成像 (D) 的低密度(蓝色箭头);E和F:胼胝体和幕上白质中流体衰减反转恢复序列的高信号(蓝色箭头);G:CT血管造影未见明显异常;H:第4天的增强MRI显示病灶未强化。
同上一例:图 2 扩散加权成像的动态变化。 AD:第2天弥散加权成像(DWI)显示胼胝体(AC)和双侧半卵圆中心(BD)有明显的异常受限弥散(蓝色箭头);EH:第4天DWI显示弥散受限区域明显(F和G)但未减少(蓝色箭头);IL:出院后1.5个月DWI显示病灶消失。
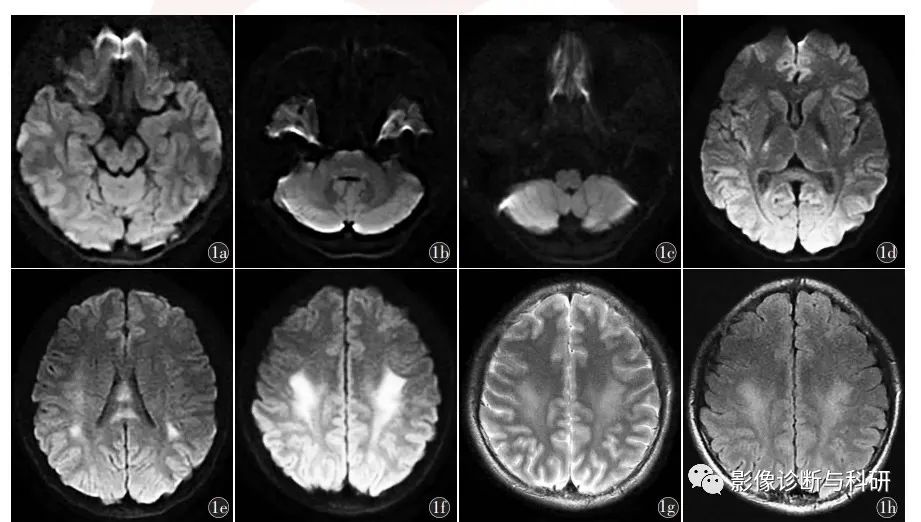
男,15岁,因“发作性言语不清2 d,症状加重3 h”,双侧面瘫及构音障碍,四肢肌力Ⅴ -级。11 月27 日行头部MR,脑干未见明显异常(图1a ~ 1c),扩散加权成像(DWI)示双侧内囊后肢、胼胝体、半卵圆中心多发对称性病灶,弥散受限(图1d ~1f),T2加权成像(T2 weighted imaging,T2WI)及液体衰减反转恢复(FLAIR)序列示双侧半卵圆中心稍高信号(图1g,1h)。
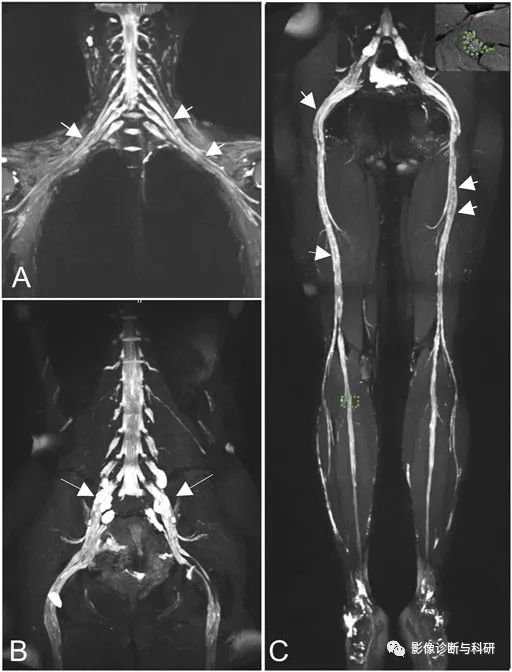
图 2。冠状磁共振神经成像(MRN)显示臂丛神经(A)弥漫性均匀肥大和腰骶神经丛(LS)多灶性梭状肥大[ (B),长箭头)],信号强度增加。神经干在臂神经、LS 神经丛和坐骨神经中表现出虫蛀信号减少(短箭头)。右腓神经感兴趣区域的放大视图显示在图像的右上角(C)。
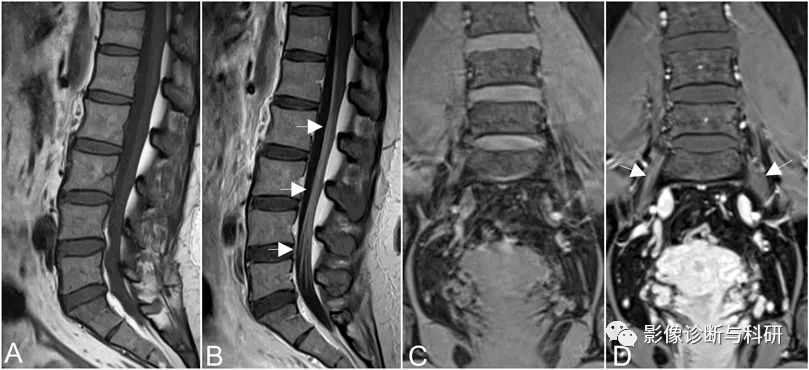
图 3。钆给药后,在T1加权图像上观察到马尾明显增强[ (A,B)中的箭头],肥厚神经丛的神经根( (C,D)中的箭头]有轻度增强。
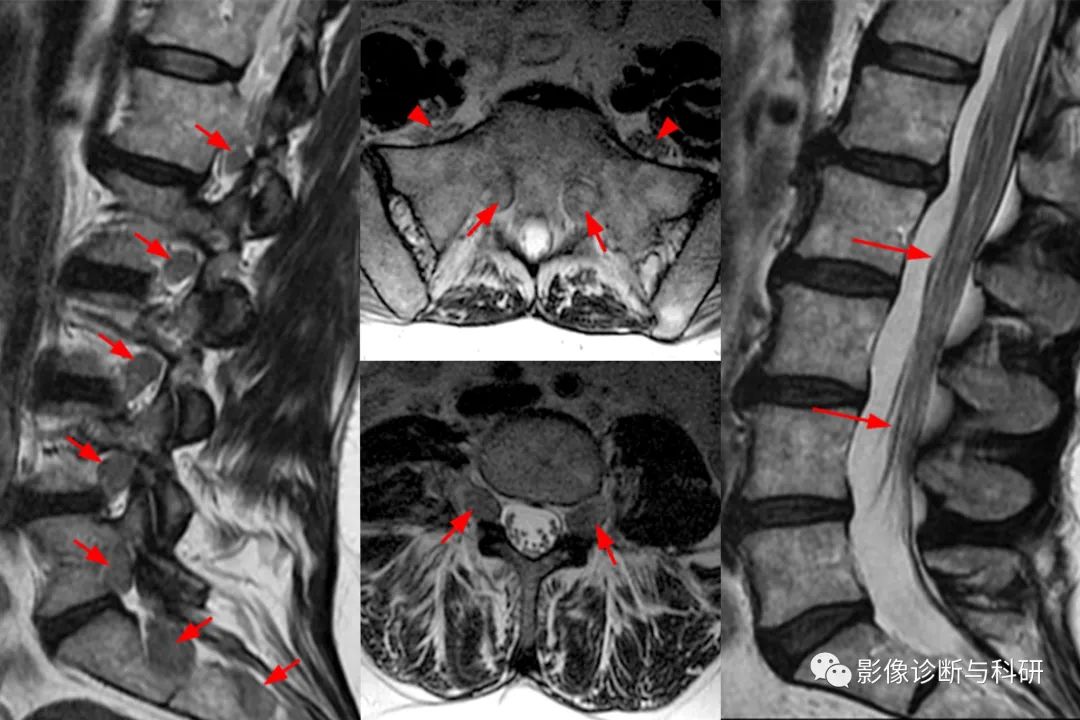
矢状位 T2 加权图像 (2a) 显示增厚的马尾神经根(箭头),旁矢状位 T2 加权图像 (2b) 显示增大的 L1-5 和 S1-2 神经根(短箭头)。图 2c,在 L4-5 水平看到放大的双侧 L4 神经根(短箭头)。图 2d 显示神经孔内放大的双侧 S1 骶神经根(短箭头)。还要注意骶翼前方突出的左右 L5 椎间孔外神经根(箭头)。
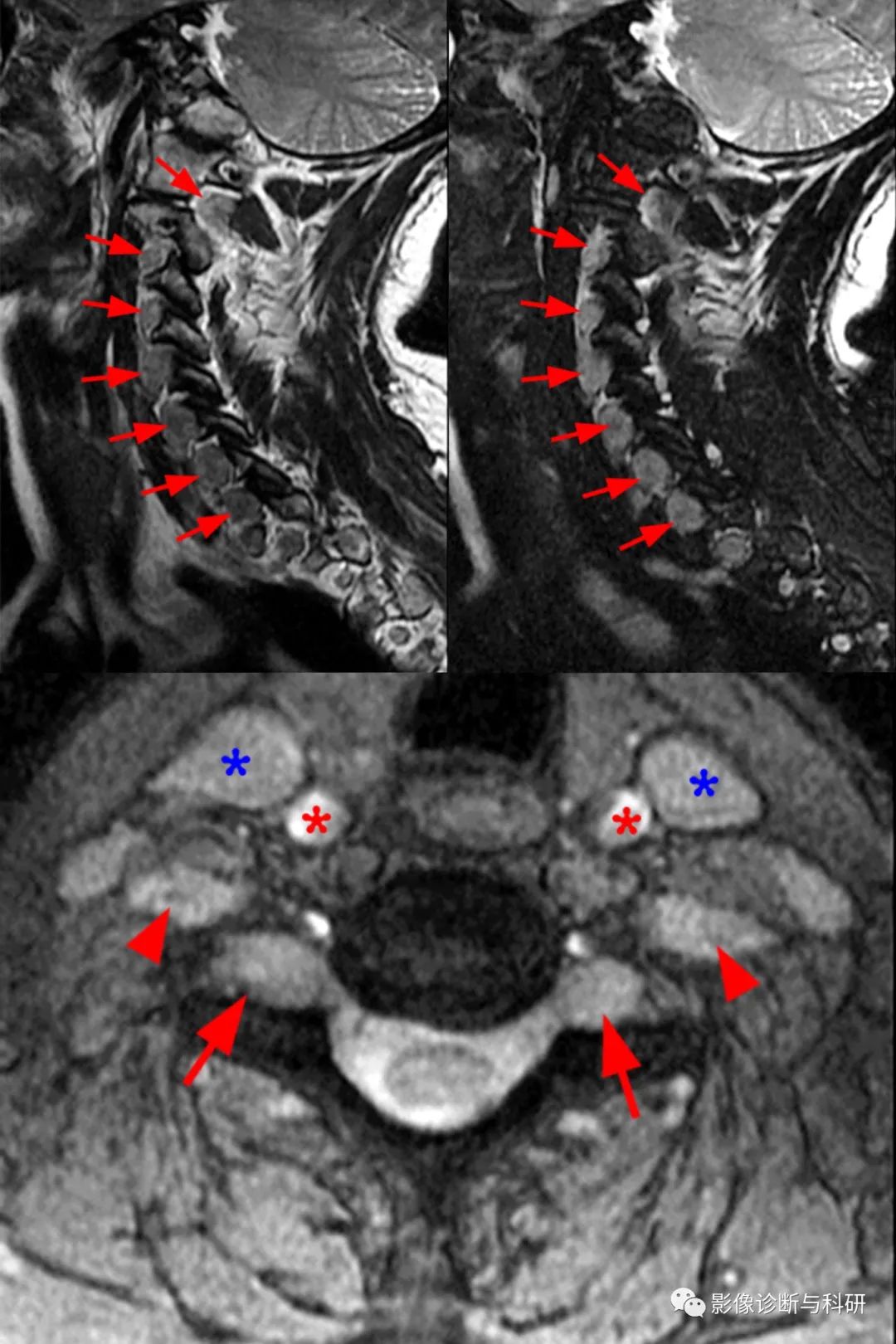
测试病例患者颈椎的旁矢状位 T2 加权 (3a) 和旁矢状位 T2 加权脂肪抑制 (3b) 图像显示 C2-T1 神经根增大(短箭头)。C6-7 水平的轴向 2D MERGE 序列 (3c) 显示背根神经节处增大的双侧 C7 神经根(短箭头)和增大的双侧孔外 C6 神经根(箭头)。颈内静脉(蓝色星号)和颈总动脉(红色星号)也可以在此水平看到。
鉴别诊断 CMT主要需要与一些累及周围神经的其他遗传性疾病相鉴别,如Krabbe脑白质营养不良、异染性脑白质营养不良、线粒体病、遗传性痉挛性截瘫和遗传性共济失调等。它们除具有周围神经病以外,还有神经系统其他部位和非神经组织器官受累的表现,CMT较少有周围神经以外的其他系统受累,肌电图及肌肉活检病理及基因检测有助于鉴别诊断。
作者:影像诊断与科研
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







腓骨肌萎缩症(CMT),又称遗传性运动感觉神经病
34