
JAMA子刊:华山医院团队发现:Batoclimab在重症肌无力患者中的有效性和安全性
2024-04-09 MedSci原创 MedSci原创
在成年全身型重症肌无力患者中,batoclimab显著提高了MG-ADL评分的持续改善,同时患者对其的耐受性良好。
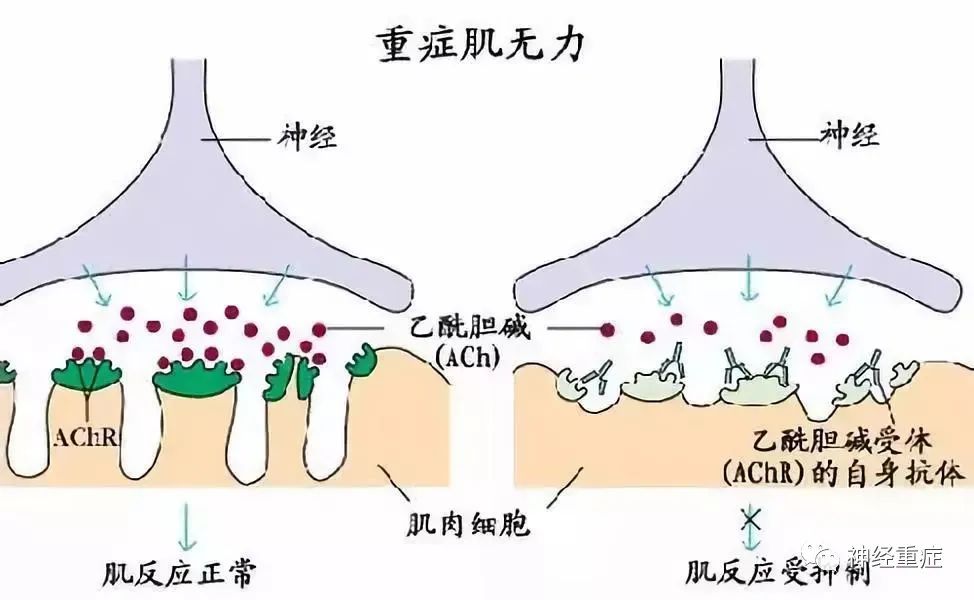
重症肌无力是一种以骨骼肌无力为特征的慢性自身免疫性神经肌肉疾病,其核心机制在于自身抗体对神经肌肉接头的破坏。为了寻求更有效的治疗方法,研究人员发现新生儿片段结晶化受体(FcRn)拮抗剂efgartigimod和rozanolixizumab具有显著潜力。这两种拮抗剂均通过降低血液循环中的免疫球蛋白G(IgG)水平,成功减轻了全身型重症肌无力患者的症状,为这一疾病的治疗开辟了新的途径。
在这一背景下,来自复旦大学附属华山医院神经内科赵重波教授及其团队开展了Batoclimab的一项III期临床研究,旨在评估单克隆IgG1抗体batoclimab在全身型重症肌无力患者中的疗效和安全性,验证batoclimab是否能够成为治疗重症肌无力的有效药物,以期为患者提供更安全、更可靠的治疗选择。其研究结果发表于JAMA Neurology。

图1:研究发表于JAMA[1]
这项由中国27家研究中心共同参与开展的随机、双盲、安慰剂对照平行研究,在2021年9月15日至2022年6月29日期间共纳入132例全身型重症肌无力成人患者,其中131例为乙酰胆碱受体/肌肉特异性受体酪氨酸激酶(AChR/MuSK)抗体阳性患者。
这些患者随机分配至Batoclimab治疗组和安慰剂组,进行以6周为一个周期的皮下注射治疗,以患者重症肌无力日常生活量表(MG-ADL)得分较基线的变化情况,评估Batoclimab治疗对疾病症状的持续改善效果。
结果显示(如图2),在治疗开始的第二周,batoclimab治疗组患者MG-ADL评分改善率曲线就与对照组产生了明显的分离,这提示batoclimab治疗能够快速起效,发挥效果。
此外,在第43天,结束了第一个治疗周期后,batoclimab治疗组的ADL评分持续改善率(ADL评分较基线改善3分且连续持续4周的比例)达58.2%(39/67),显著高于对照组(31.3%,20/64),提示batoclimab治疗能够显著改善患者症状,且具有可持续的治疗效果。
值得注意的是,在评估治疗相关的不良事件(AEs)时,安慰剂组的发生率为36.9%(24/65),而batoclimab组的发生率为70.1%(47/67),关于严重不良事件(SAEs)的发生率,安慰剂组为7.7%(5/65);batoclimab组为3.0%(2/67)。
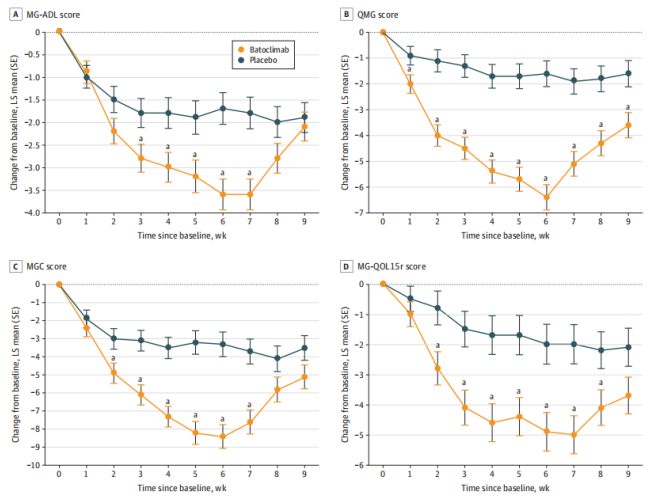
图2:第一个治疗周期抗体阳性患者MG-ADL、定量重症肌无力(QMG)、复合重症肌无力(MGC)和MG生活质量15-修订(MG-QOL15r)评分的时间特征[1]
根据上述结果,我们可以明确得出:在成年全身型重症肌无力患者中,batoclimab显著提高了MG-ADL评分的持续改善,同时患者对其的耐受性良好。其临床疗效以及IgG降低程度与先前报道的efgartigimod和rozanolixizumab药物相当。然而,为了进一步了解batoclimab的安全性,未来需要开展更大样本量的研究。
此外,鉴于batoclimab的潜在疗效及其可能引发的不良反应,未来的研究应当进一步探索batoclimab与其他治疗方法的联合应用,以寻求最优化的治疗方案并最大化患者的治疗效益。
同时,针对特定患者群体(例如老年人、儿童或患有其他疾病的患者)的研究也显得尤为关键,这将有助于我们更全面地评估batoclimab在不同患者群体中的安全性和有效性,从而为临床决策提供更为精准的依据。
原始出处:
Yan C, Yue Y, Guan Y, et al. Batoclimab vs Placebo for Generalized Myasthenia Gravis: A Randomized Clinical Trial. JAMA Neurol. Published online March 04, 2024.
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#重症肌无力# #batoclimab#
16